Capítulo 36
Urología pediátrica
- Dra. Carolina Acuña M. (1), Dr. Pedro José López E. (2), Dra. Francisca Yankovic B. (3)
- (1) Hospital Padre Hurtado – Clínica Alemana – (2) Hospital Exequiel González Cortés – Clínica Alemana – (3) Hospital Exequiel González Cortés – Clínica Santa María
Genitales externos
Testículo no descendido
El testículo no descendido corresponde a una de las malformaciones genitales más frecuente en niños. Su incidencia en recién nacidos de término es de 1,5 – 4% y en prematuros puede llegar a 45%(1,2). Durante los primeros meses post parto puede existir un descenso testicular espontáneo. Este descenso generalmente ocurre en los primeros tres meses y es secundario al estímulo hormonal natural que existe en este período, conocido como “mini pubertad”. Por esta razón, en pacientes de término y tras cumplir 6 a 12 meses de vida, la incidencia baja al 1%. La clasificación más utilizada actualmente es aquella que divide los testes no descendidos en palpables (80%) y no palpables (20%)2. La figura 1 resume las principales formas de presentación del testículo no descendido.

Definiciones
Criptorquidea es aquel teste que se encuentra en algún lugar del trayecto normal del descenso testicular.
Ectopia testicular es aquel teste que se encuentra fuera del trayecto normal del descenso testicular. La ectopia más frecuente es inguinal supra-aponeurótico. Otras posibilidades son femoral, perineal y púbico entre otros.
Teste retráctil (en ascensor), es aquel teste que baja fácilmente al escroto, es decir, sin tensión del cordón espermático, permanece ahí, y debido a un reflejo cremastérico intenso e hiperactivo, sube a canal inguinal. La diferencia con un teste criptorquídico en canal inguinal, es que, al bajarlo al escroto, el teste se queda abajo sin subir rápidamente.
Teste no palpable es aquel teste que no se palpa en canal inguinal tras ser examinado en las mejores condiciones posibles, con un paciente tranquilo (sin llorar), ambiente cálido (el frio asciende los testes) y por un urólogo pediátrico con experiencia.
La importancia del diagnóstico oportuno de los testículos no descendidos reside en que éstos desde los 6 meses de vida pueden experimentar cambios estructurales condicionados por el ambiente inadecuado en que se desarrollan. A esa edad podrían aparecer cambios histológicos iniciales, donde hay una degeneración de células germinales que aumenta con la edad; de un 1% en casos normales, llega a un 20% en criptorquídea a los 2 años, 40% a los 6 años y 45% a los 10 años2; generando espermatogonias incapaces de completar su proceso habitual de maduración. Este fenómeno podría llevar a que en la pre-pubertad y adolescencia se generen focos de displasia celular, con en consecuente incremento del riesgo de cáncer testicular, siendo 3 a 10 veces más frecuente en hombres con antecedente de criptorquídea.
Tratamiento
Dado que el daño a nivel histológico puede comenzar a los 6 meses, es recomendable derivar a esta edad para la resolución quirúrgica y evitar postergar la cirugía.
El tratamiento de elección del testículo no descendido es la cirugía:
Testículo palpable: en los casos donde el testículo es palpable en el canal inguinal, ectópico o se trata de un teste retráctil con disminución del crecimiento testicular, se realiza liberación de los elementos del gubernáculum, cierre de hernias en los casos que existan, disección hacia el retroperitoneo y orquidopexia sin tensión al escroto (pexia o bolsillo tipo surraco).
Testículo no palpable: El tratamiento depende si se trata de un testículo no palpable que se encuentra en el canal inguinal, en cuyo caso se puede proceder de la misma manera que en el caso anterior (testículo palpable). Si el testículo no fue encontrado en el examen bajo anestesia y/o en la ecografía, se requiere de una laparoscopía exploradora. En estos casos al ingresar a la cavidad abdominal existen 4 posibilidades:
– Testículo intraabdominal viable (40%).
– Testículo ausente y vasos sanguíneos ciegos a nivel del anillo inguinal profundo (20%).
– Testículo atrófico (evanescente) inguinal: vasos sanguíneos y deferente ingresando por canal inguinal con conducto inguinal profundo generalmente cerrado (30%).
– Testículo normal en el canal inguinal, vasos sanguíneos y deferente ingresando por canal inguinal con conducto inguinal profundo generalmente abierto (10%).
La técnica quirúrgica va a depender del hallazgo intraoperatorio y de las características del testículo. En aquellos testículos de buen aspecto se puede realizar el descenso testicular y orquidopexia en un tiempo. Esto consiste en liberar los vasos espermáticos y los elementos del gubernáculum permitiendo la llegada del testículo al canal inguinal. Esta técnica es más utilizada en los testículos no palpables tipo “peeping”, donde el teste esta a nivel del anillo inguinal profundo, entrando y saliendo de este. También se puede hacer descenso y orquidopexia en dos tiempos (Fowler-Stephens), donde en el primer tiempo se seccionan los vasos espermáticos, esperando que, en un periodo mínimo de 6 meses, la vascularización vía arteria deferencial del teste permita su descenso sin problemas. El riesgo de atrofia testicular con esta técnica es de aproximadamente un 10-15%1. La figura 2 resume las opciones terapéuticas en el manejo del testículo no descendido.

Hipospadia
Definición
La hipospadia es un defecto congénito del pene, caracterizado por una asociación de tres variaciones anatómicas de los genitales externos: desembocadura ventral del meato uretral (entre glande y periné), prepucio alado (ausencia de unión hacia ventral) y curvatura peneana ventral (cuerda/chordee). Estos tres componentes no siempre están presentes en forma concomitante. También se puede definir a la hipospadia como una hipoplasia de los tejidos que forman el aspecto ventral del pene, más allá de la división del cuerpo esponjoso.
La incidencia es de 1 en 250 a 300 recién nacidos vivos. Si bien no es clara su etología, los posibles factores etiológicos son: endocrinopatías, bajo peso al nacer, edad materna avanzada (probablemente por insuficiencia placentaria), antecedentes de diabetes materna, y exposición a factores “disruptores” ambientales (pesticidas, tóxicos, productos químicos industriales, etc.)1. Actualmente, se ha avanzado en la búsqueda de alteraciones en genes específicos que se asocien con la aparición de hipospadia y otras variaciones del desarrollo sexual. En esta línea se ha encontrado que la alteración en el gen kappa diacilglicol quinasa (DGKK) puede estar presente en casi el 60% de las hipospadias proximales1. También se ha planteado la posibilidad de una herencia poligénica; la incidencia en parientes de primer grado de pacientes afectados es de 7 a 10%, y de hasta 10 – 20% en formas graves. Es muy importante destacar, que formas severas de hipospadias y/o cuando están en asociación con otra alteración genital, como un testículo no descendido, pueden estar en el contexto de un desorden del desarrollo sexual (DDS), por lo que se aconseja sospechar esta condición y derivarla en forma oportuna3. La asociación con alteraciones en el tracto urinario es infrecuente en hipospadias distales aisladas, pero aumenta su incidencia en hipospadias proximales.
Clasificación
Existen varias clasificaciones de hipospadias, pero la más utilizada está determinada por la localización del meato uretral el cual puede cambiar su posición inicial después de liberar la cuerda peneana al momento de la cirugía.
En la tabla 1 se muestran la clasificación de las hipospadias se acuerdo a la posición del meato y el porcentaje de presentación de ellos. Sin embargo, en términos prácticos se dividen en distales y proximales
| DISTAL | Glandular
Coronal Subcoronal |
70 % |
| MEDIAL | 1/3 anterior
1/3 medio 1/3 proximal |
20 % |
| PROXIMAL | Peno-escrotal
Escrotal Perineal |
10% |
Tabla 1. Clasificación de las hipospadias y porcentaje de presentación.
Cuadro Clínico y diagnóstico
El diagnóstico es clínico durante la exploración física genital. Sin embargo, en ocasiones cuando el prepucio está intacto puede pasar desapercibida los primeros meses de vida. En caso de tener una hipospadia muy distal, que puede parecer un meato normal, el diagnóstico se puede hacer por la presencia de prepucio alado, con o sin curvatura peneana.
Las hipospadias más complejas hay que distinguirlas de un recién nacido con Desordenes del Desarrollo Sexual (DDS). Esta patología no se manifiesta con una sintomatología específica, no tiene mayor riesgo de ITU ni ocasiona obstrucción urinaria.
Tratamiento
El tratamiento de las hipospadias es quirúrgico, que preferentemente se debe llevar a cabo entre los 6 y 18 meses de edad, después de la elevación hormonal que se presenta en los varones entre los 2 y 3 meses de edad (minipubertad). Es ideal realizarlo antes al retiro de pañal, pues facilita el manejo post operatorio. Los objetivos de la intervención (en orden de importancia) son: corregir la curvatura peneana, lograr un meato uretral normotópico y un glande cónico; con una uretra de buen calibre y, por último, una cobertura cutánea adecuada. En ocasiones, cuando el pene es muy pequeño (menor que el percentil 3 o por debajo de 2 desviaciones estándar para el tamaño normal para la edad), el paciente debe ser evaluado por un endocrinólogo, para administración de testosterona, con el objetivo de estimular el crecimiento peneano. Del mismo modo, otro índice para determinar si el tamaño del pene permitiría un éxito quirúrgico adecuado, el glande en su parte transversal mas ancha debe tener no menos de 14mm.
Existen múltiples técnicas para la corrección de hipospadias, las cuales han cambiado a lo largo de los años, lo que implícitamente habla que no existe aun una técnica ideal para la corrección de esta condición. La uretroplastía se puede dividir en cirugías de “una etapa” o “en etapas”; dependiendo del tipo y grado de hipospadias. La reparación en etapas se utiliza en hipospadias severas, se basa en que durante el primer tiempo se corrige la curvatura, reposicionando el meato uretral más proximal, además de colocar un injerto libre de piel prepucial en la cara ventral para la reconstrucción uretral en una etapa posterior. En caso de no disponer piel prepucial se pueden utilizar otros injertos libres; como el de mucosa oral, ya sea de labio inferior o de mejilla. La segunda etapa se realiza 6 meses después; reconstruyendo uretra empleando injerto colocado en la primera etapa. La tabla 2 resume los tipos de cirugías recomendados por tipo de hipospadias.
| DISTAL | MAGPI
Mathieu TIP |
| MEDIAL | Mathieu
Onlay flap TIP En etapas |
| PROXIMAL | En etapas |
Tabla 2. Tipos de cirugías de corrección de hipospadias dependiendo de grado.
Complicaciones
Pre-operatorias: Durante la infancia, la hipospadias no se presenta con complicaciones. En caso de no ser corregida quirúrgicamente, las hipospadias pueden presentar falta de control del chorro miccional (necesidad de orinar sentado) y/o problemas para tener relaciones sexuales.
Post-operatorias:
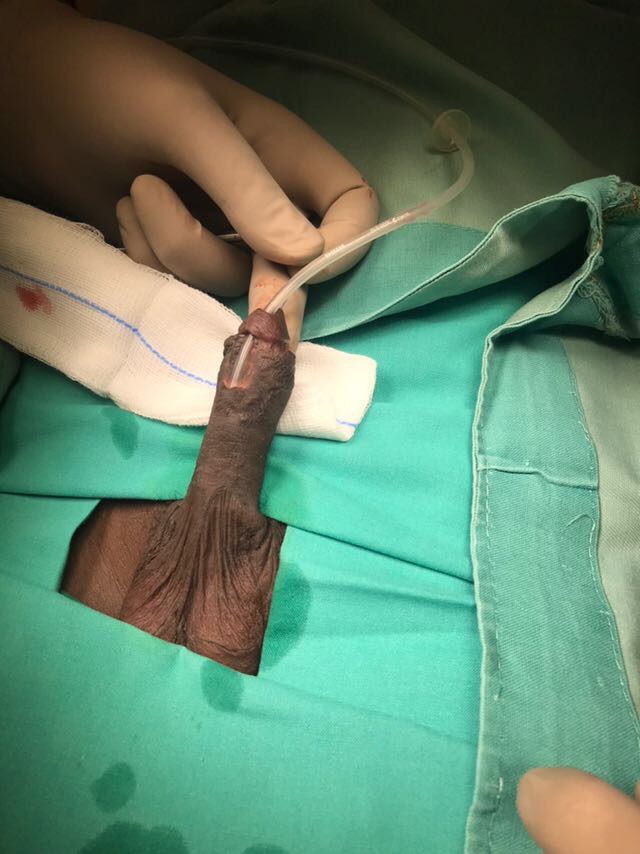
- Fístula uretrocutánea: complicación más frecuente, aproximadamente en un 5-15% de los pacientes con hipospadia distal y hasta en un 50% de los pacientes con hipospadia proximal. Pueden asociarse a estenosis y/o divertículo uretral. Se sugiere que la reparación de la fístula se realice mínimo 6 meses después de la cirugía. La figura 3 muestra un paciente post operado con una fistula uretrocutanea post operatoria.
- Estenosis: la más frecuente es la meatal, pero también puede darse en la posición proximal en la unión entre el meato original del paciente y la uretroplastia. Se evidencia con una disminución en el chorro miccional, latencia al orinar o dificultad al vaciamiento vesical. Generalmente requiere de corrección quirúrgica con una meatotomía y/o re-hacer la ureteroplastía.
- Dehiscencia: generalmente del glande, requiere corrección quirúrgica.
- Divertículo uretral: está más relacionado a un procedimiento de colgajo en isla y/o una estenosis, ya que el chorro miccional produce presión dentro del colgajo que, al ser un tejido expandible, la que va formando un divertículo. Se diagnostica con visión de balonamiento de la uretra, con goteo postmiccional y/o ITU.
- Cuerda persistente: asociada frecuentemente a una liberación insuficiente de la misma, sin embargo, puede ser secundaria a fibrosis o hipoplasia residual. Requiere tratamiento quirúrgico para su corrección, que por puede ser en etapas.
- Uretra pilosa: se presentaba antiguamente cuando se utilizaban colgajos prepuciales-escrotales (colgajo en isla), que, al tener folículos pilosos, con la pubertad se desarrollaban pelos en el interior de la uretra lo que provoca litiasis y/o ITU.
Pronóstico
Es generalmente bueno, ya que frecuentemente se alcanzan los objetivos de la cirugía, con una estética adecuada y una buena función miccional y sexual. El seguimiento postoperatorio debe de hacerse a largo plazo, idealmente hasta la adolescencia.
Desordenes del desarrollo sexual (DDS)
El término DDS (en inglés DSD Disorders of Sexual Differenciation) hace referencia a un grupo heterogéneo de condiciones congénitas que pueden afectar la determinación y/o diferenciación sexual del paciente, produciéndose un desarrollo sexual anatómico, gonadal y/o cromosómico no concordante o atípico (3,4). Los pacientes con DDS requieren de un manejo multidisciplinario y en etapas. En el periodo de recién nacido debe evaluarse el diagnóstico, la explicación-asimilación de los padres, la asignación de género. Dependiendo del paciente se podrá incluir el manejo médico de las alteraciones metabólicas, y ocasionalmente el tratamiento quirúrgico reconstructivo, sin olvidar apoyo psicológico y el acompañamiento de la familia5. Es fundamental estandarizar el manejo y estudio de estos pacientes en los distintos hospitales del país de tal manera de disminuir procedimientos controversiales y tomar decisiones basados en consensos, evidencia y experiencia.
Clasificación3
| Cromosoma Sexual | 46 XY | 46 XX |
| 45 X0 (Sd. Turner) | Alt. Desarrollo testicular.
(Disgenesia gonadal completa y parcial) |
Alt. Desarrollo ovárico (Disgenesia Gonadal) |
| 47 XXY (Sd. Klinefelter) | Alt. De la síntesis y/o acción de andrógenos (Deficiencia 17 HDH, % -reductasa, CASI, PAIS) | Exceso de andrógenos (HSRC, deficiencia de aromatasa) |
| Disgenesia Gonadal Mixta (45 X/46 XY)
Quimera (46 XX/46 XY) |
Sd. Persistencia de conductos Mullerianos (mutación o deficiencia de hormona antimüleriana) | Miscelanios Extrofia Cloacal, atresia vaginal. |
| Miscelanias (hipospadias, epispadias, extrofia) |
Evaluación Clínica en recién nacido
Anamnesis Materna: debe incluir algunos aspectos fundamentales que orientan al diagnóstico como el uso de drogas u hormonas durante el embarazo, antecedentes de muerte fetal o recién nacido con crisis adrenogenital (deshidratación, hiperkalemia e hiponatremia), historia materna de virilización, consanguinidad de padres y/o historia familiar de DDS
Examen físico: Esta es la clave frente la sospecha de DDS, en el recién nacido se deben evaluar los siguientes hallazgos al examen físico:
- Gónadas, ubicación y consistencia
- Medición de falo
- Ubicación del meato uretral
- Morfología de pliegues labio-escrotales
- Distancia entre ano y tubérculo genital (uretra): mientras más larga es signo de androgenización
Los hallazgos al examen físico del recién nacido que sugieren trastornos del desarrollo sexual son:
- Aparentes genitales femeninos con clitorimegalia, fusión de los labios mayores, y masas inguinales o labiales.
- Aparentes genitales masculinos con testículos no palpable bilaterales
- Hipospadia perineal.
- Hipospadia (distal o próximal) con testículos no descendidos uni o bilateral.
- Seno urogenital.
- Recién nacido con crisis adreno-genital o HTA.
- Hiperpigmentación areolar o labio-escrotal.
- Alteración genital/urológica manifiesta como en los niños con extrofia cloacal.
La sospecha de DDS después del periodo de recién nacido se puede orientar debido a:
- Pubertad atrasada o incompleta
- Virilización en niñas
- Amenorrea Primaria
- Ginecomastia en varones
- Gónada indiferenciada en descenso testicular en paciente asignado masculino
- Hematuria en varones cíclica y macroscópica
- Crisis Adrenal
- Remanentes müllerianos en laparoscopía
- Hematocolpos/ dolor cíclico
- Tumores gonadales (gonadoblastoma)
- Niña con hernia inguinal
- Infertilidad
Laboratorio y estudio inicial
Ante la sospecha de DDS es vital descartar la Hiperplasia Suprarrenal congénita, que es la única entidad que en este periodo puede ser fatal por sus desequilibrios metabólicos y electrolíticos. Para eso el estudio inicial debe incluir; Electrolitos plasmáticos, Hemograma, glicemia y renina plasmática, FISH SRY – Cariotipo, Estudio Hormonal Plasmático al 3-4° día de vida: 17 Hidroxi-progesterona, Testosterona y DHT, Esteroides urinarios. La ecografía evalúa la presencia de genitales internos, vía urinaria y riñones, suprarrenales y columna.
Equipo multidisciplinario con experiencia
Los pacientes portadores de DDS representan un desafío en su diagnóstico y tratamiento. Por lo mismo deben ser tratados en centros especializados multidisciplinarios con el fin de optimizar el manejo de estos pacientes, equipos que deben integrar desde el inicio a los padres en las decisiones.
Frente a la asignación de sexo y antes de la tomar una decisión, se debe evaluar el diagnóstico, apariencia de los genitales, opciones quirúrgicas de corrección requiriendo la mínima cantidad de procedimientos, necesidad de terapia hormonal a permanencia, potencialidad de fertilidad, opinión de la familia, aspectos socioculturales y potencial riesgo de malignidad. Sabemos que la asignación de género ha sido exitosa cuando se logra una apariencia acorde al género con la menor cantidad de procedimientos, una función sexual y reproductiva adecuada y la estabilidad psicosocial. Es primordial establecer equipos multidisciplinarios que incluyan a la familia y realizar un seguimiento sistemático y estructurado siempre enfocado hacia lograr una persona integrada y feliz consigo mismo y con /en la sociedad
Complejo extrofia vesical / cloacal
El complejo cloaca – extrofia – epispadias, es un amplio espectro de anomalías congénitas que se debe al ser resultado de un mismo defecto embriológico, pudiendo abarcar desde la epispadia glandular hasta la extrofia cloacal.
Embriología
La separación de la cloaca primitiva en el seno urogenital y el intestino posterior ocurre durante el primer trimestre del embarazo, casi al mismo tiempo en que se constituye la pared abdominal anterior. Una falla en la migración de las células mesenquimáticas entre las capas ectodérmica y endodérmica de la pared abdominal inferior provoca inestabilidad de la membrana cloacal. La rotura prematura de esta membrana antes de su migración caudal conduce al desarrollo de este conjunto de anomalías infraumbilicales. Si la rotura ocurre después de la separación completa de los tractos genitourinario y gastrointestinal se produce una extrofia vesical clásica. Sin embargo, si esta ocurre antes del descenso del tabique urorrectal se produce externalización del tracto urinario inferior y de la porción distal del tracto gastrointestinal, dando lugar a una extrofia de cloaca(3,6).
Epidemiología
El complejo extrofia vesical – cloacal afecta con mayor frecuencia a personas de raza blanca. La extrofia vesical clásica es el subtipo predominante (50%), presentándose en 3,3/100.000 nacidos vivos, y la extrofia cloacal se ve en 1/200.000. La extrofia vesical clásica es más frecuente en hombres (3:1). La extrofia de cloaca es igual en ambos sexos (1:1). La mortalidad en pacientes con extrofia vesical es baja (4%). La sobrevida de niños con extrofia cloacal ha mejorado de 50% en 1960 hasta 80% en la actualidad7.
Manifestaciones clínicas
Período fetal: En el embarazo es posible sospechar esta alteración al existir imposibilidad de identificar la vejiga en ecografías sucesivas, disminución del espesor de la pared abdominal, cordón umbilical de implantación baja, genitales de posición anormal (anterior o posterior), falo corto, aumento del diámetro pélvico, con separación de las ramas del pubis, así como onfalocele, malformaciones de las extremidades inferiores y/o mielomeningocele (sugerentes de extrofia cloacal).
Período de RN: La mayoría de estas variantes son fácilmente identificables al nacer. Por lo general, la extrofia vesical se ve más recién nacidos de término con buen peso de nacimiento, y los RN con extrofia cloacal suelen ser de pretérmino, con bajo peso de nacimiento
Infancia: Las variantes infrecuentes pueden pasar desapercibidas e identificarse en la infancia sólo por incontinencia urinaria persistente o alteraciones de la marcha. Especial atención en pacientes de sexo femenino con incontinencia urinaria refractaria en la que podría haber pasado desapercibida una epispadias femenina.
Examen físico
En la extrofia vesical clásica, están involucrados el tracto urinario inferior, genitales y aparato locomotor, mientras que en la extrofia de cloaca hay mayor compromiso del tracto gastrointestinal y SNC.
Extrofia vesical clásica: La pared abdominal es alargada, el ombligo es de implantación baja, se ubica en el borde superior de la placa vesical y puede asociarse a un defecto herniario o un pequeño onfalocele. La vejiga abierta anteriormente, con su mucosa totalmente expuesta. El ano está más anterior, pero la función esfinteriana es normal. Los huesos del pubis están ampliamente separados y, además pueden estar acortados y rotados externamente (30%).
Extrofia vesical femenina: El clítoris es bífido, los labios mayores están separados y la vagina es corta, y más anterior.
Extrofia vesical masculina: La placa uretral está abierta y se extiende a lo largo del falo corto, ancho y con curvatura dorsal. El glande está abierto y es plano. En ocasiones pueden existir variaciones de la extrofia clásica, como se ve en la figura 1, donde existe apertura de la placa vesical, pero con el pubis y pene intactos.
Extrofia de Cloaca: Los músculos rectos y los huesos del pubis están separados. La vejiga está abierta en la pared abdominal inferior y dividida en 2 mitades adyacentes al segmento expuesto del ciego. Los orificios que comunican el íleon terminal, el apéndice (uno o dos) y el intestino distal son evidentes al interior de la placa cecal y el Íleon terminal puede prolapsarse como un «tronco» a través de ésta. Se presenta con ano imperforado y puede asociarse a onfalocele. Un 95% presenta mielodisplasia y un 65% tiene una malformación de las extremidades inferiores. En hombres, el falo generalmente es bífido y pequeño, con cada hemiglande ubicado caudal a cada hemivejiga, o puede estar ausente. En mujeres, el clítoris es bífido y puede haber dos hemivaginas con un útero bicorne. Las figuras 4 y 5 muestran el aspecto preoperatorio de una extrofia vesical


Estudios de imágenes
Permiten identificar y diagnosticar otras posibles malformaciones tanto pélvicas, abdominales como de columna y medula vertebral. Lo básico puede ser ecografía renal, ecografía de médula espinal, radiografía de columna vertebral y de pelvis, Otros estudios más tardíos puede ser RNM y uretrocistografía.
Manejo
De existir diagnóstico antenatal, lo ideal es que pueda nacer en un centro especializado, donde se pueda hacer consejería a los padres desde el embarazo. No existe experiencia que avale la indicación de cirugía fetal. Ideal evitar el parto vaginal por existir mayor riesgo de lesión de la placa vesical. Hoy ha dejado de ser una emergencia y/o urgencia medica quirúrgica, lo que permite un trasladado post natal en buenas condiciones a un centro especializado que pueda manejar estos niños con patologías de baja frecuencia y alta complejidad7.
Tratamiento quirúrgico
Los objetivos del abordaje quirúrgico actualmente son; (1) reconstrucción de la pared abdominal, (2) reubicación y cierre anatómico de la vejiga extrófica, (3) preservar la función renal y lograr la continencia urinaria y (4) reconstrucción de los genitales externos. Históricamente, la reconstrucción se realizaba en 3 etapas comenzando en la etapa de RN. Sin embargo, en la actualidad existen equipos de expertos multicéntricos que pueden organizar la cirugía en forma diferida ofreciendo lo mejor a cada niño al acumular experiencia en este equipo en una patología de baja frecuencia y alta complejidad.
Los pasos quirúrgicos son:
a) Cierre de la vejiga, con aproximación de los huesos del pubis, sin necesidad de osteotomía. En pacientes con extrofia de cloaca debe realizarse derivación intestinal.
b) La etapa dos, consiste en el cierre completo del pene. En algunos centros realizan el procedimiento de Kelly o Reconstrucción Radical de Partes Blandas que le permite en varones darle mayor longitud al pene. Se puede intentar en esta etapa la continencia vesical
c) La tercera etapa consiste en la evaluación de la continencia urinaria que se puede mejorar con cirugía de cuello vesical y/o cistoplastía de aumento
Complicaciones
Al ser una condición compleja en sí, esta presenta sus propias complicaciones tempranas como dehiscencia de la herida operatoria, prolapso vesical, fístulas uretrales o vesicocutáneas (4 – 19%), estenosis uretral (8%). Por otro lado, las tardías pueden ser RVU, ITU recurrente, incontinencia urinaria, litiasis vesical, fibrosis de la pared abdominal, ruptura vesical, prolapso uterino, insuficiencia renal, eyaculación retrógrada, oligoespermia e infertilidad, Síndrome Intestino Corto (en Síndrome extrofia cloacal)
Pronóstico
Hoy en día, estos pacientes tienen una excelente sobrevida a largo plazo, logrando continencia urinaria hasta en 70% de los pacientes con extrofia vesical y algo menor en casos de extrofia cloacal (50%), requiriendo derivaciones urinarias e intestinales permanentes en muchos. En general, la función sexual está conservada y la mayoría son fértiles. En las mujeres con esta condición, se recomienda el parto por cesárea, para evitar daños en los mecanismos de continencia
Obstrucción del sistema urinario
Diagnóstico y Manejo Perinatal1
Las anomalías congénitas de vías urinarias y riñón constituyen el 20 a 30% de todas las anomalías diagnosticadas durante el periodo prenatal. La dilatación del tracto urinario es una condición que debe estudiarse desde su diagnostico prenatal. La más frecuente es la hidronefrosis (HN) que corresponde a la dilatación de la pelvis renal con o sin dilatación de los cálices. La mayoría de las veces esta dilatación de la pelvis renal es transitoria y fisiológica. Sin embargo, en un porcentaje menor, la presencia de HN será consecuencia de una alteración patológica del tracto urinario tales como la obstrucción pielo ureteral o el reflujo vésico ureteral, entre otras. Estas condiciones pueden o no asociarse a compromiso de la función renal. No existe conceso con respecto a cuál es el mejor sistema de evaluación – graduación de la HN para orientar posible compromiso de función renal y/o pronóstico en el período post natal. El diámetro de pelvis renal es la clasificación más utilizada, la cual mide el diámetro anteroposterior transverso (DAP) máximo de la pelvis renal.
Cuando se realiza una ecografía prenatal, los valores utilizados son los siguientes:
- DAP de 4mm es el corte riesgo bajo para HN en el segundo trimestre (<33 semanas). DAP > 4mm en esta etapa estaría asociado con un riesgo aumentado de anomalías congénitas del riñón y tracto urinario.
- DAP > a 7mm durante el tercer trimestre (>33 semanas) tienen mayor riesgo de presentarse con anomalías congénitas del riñón y tracto urinario, estas aumentan con la severidad de la persistencia de la HN fetal.
Luego de nacer, lo ideal esperar los primeros 2-3 días que pase la oliguria fisiológica para realizar una ecografía postnatal. En este examen el DAP se puede utilizar como factor estimativo. A pesar de que la mayoría de las dilataciones moderadas se van a resolver solas y no van a tener una repercusión en la función renal hay algunas que requieren de una intervención postnatal.
Etiología
HN fetal puede desarrollarse secundario a:
- Dilatación transitoria del sistema colector, siendo la causa más frecuente representa el 40-90% de las HN. Esta se produce por una estrechez transitoria de la unión pieloureteral que resuelva espontáneamente con el desarrollo.
- Uropatía obstructiva del tracto urinario alto o bajo
- Obstrucción de la unión pieloureteral, segunda causa más frecuente de HN.
- Megaureter Obstructivo primario
- Valvas de uretra posterior (VUP)
- Vejiga Neurogénica
- Otros: atresia de uretra, Síndrome Prunne Belly, etc
- Procesos no obstructivos:
- RVU, tercera causa más frecuente
Diagnóstico
El diagnostico se realiza a través de la ecografía prenatal, ésta puede determinar la necesidad de estudios postnatales como también para orientarnos a la etiología. Los factores a evaluar son los siguientes:
- Lateralidad: Al ser bilateral aumenta el riesgo de compromiso de la función renal global del paciente, oligohidroamnios, parto prematuro, etc.
- Uréter: la dilatación del uréter (hidroureteronefrosis) puede ser debido a RVU, uropatía obstructiva distal en el uréter o valvas de uretra posterior (VUP), entre otros
- Parénquima renal: adelgazamiento del parénquima o quistes corticales indican injuria o inadecuado desarrollo de la corteza renal.
- Vejiga: anormalidades como megavejiga y/o engrosamiento de la pared son consistentes con uropatía obstructiva distal a la vejiga (ej, VUP). La dilatación de la uretra proximal también es un signo de VUP.
- Volumen de líquido amniótico: oligohidroamnios es consistente con falla renal severa.
- Urinoma: secundarios a obstrucción como VUP u obstrucción de unión pieloureteral. En el 80% de los casos está asociado a un riñón displásico ipsilateral.
- Ascitis urinaria: debido a una ruptura vesical y caliceal espontanea o iatrogénica secundario a obstrucción distal o infrecuentemente a una vejiga neurogénica.
Tratamiento
La intervención fetal, como el Shunt aumenta la cantidad de líquido amniótico por lo tanto mejoran el desarrollo pulmonar y por consecuencia la sobrevida. Pero no hay claridad si la intervención fetal mejora la función renal a largo plazo. Se ha visto una alta tasa de fallecimiento y falla renal crónica en estos pacientes, requiriendo terapia de reemplazo en 2/3 de estos.
Manejo Postnatal: El objetivo principal del manejo postnatal de pacientes diagnostico de dilatación del tracto urinario prenatal es identificar anomalías congénitas del tracto genitourinario significativas. De esta manera se busca evitar estudios innecesarios como también identificar a los pacientes que requieren intervención temprano y así reducir el daño renal causado por éstas. El manejo se decide según la presencia de factores predictivos en las imágenes; principalmente por el compromiso bilateral y la severidad de la de la dilatación post natal persistente.
Al examen físico es importante estar atentos a:
- Masa abdominal: puede representar un riñón hipertrófico, secundario a una uropatía obstructiva o riñón displásico multiquístico, poco frecuente
- Vejiga palpable en hombres: signo de VUP y/o vejiga neurogénica
- Deficiencia de musculatura abdominal y criptorquidia son signos del Síndrome de Prune Belly.
- Arteria umbilical única: asociada con un mayor riesgo de anomalías del tracto urinario especialmente con RVU.
- Anormalidades de extremidades inferiores o espinales pueden sugerir una vejiga neurogénica la cual se puede presentar con HN y dilatación de uréteres.
El estudio debe realizarse con:
Ecografia postnatal: Como se mencionó, debe realizarse después de las 48hrs de vida debido a la oliguria fisiológica del RN que puede subestimar el grado de dilatación.
En este examen el DAP se puede utilizar como factor estimativo donde;
- DAP de <15 mm es poco probable que requiere tratamiento quirúrgico
- DAP entre 15 y 30mm, se realiza seguimiento, que pudiera requerir cirugía
- DAP de >30 mm es altamente probable que requiera tratamiento quirúrgico
Los pacientes con HN bilateral, con riñón hidronefrótico único o sospecha de obstrucción de vía urinaria baja (HN bilateral, engrosamiento vesical y dilatación de uréteres) requieren de estudio urgente dentro de 48hrs desde el nacimiento por el riesgo aumentado de tener una enfermedad significativa y necesidad de manejo precoz.
Uretrocistografía miccional retrógrada: Debe indicarse racionalmente en casos seleccionados de pacientes con diagnóstico antenatal de HN. Principalmente indicado en forma precoz ante la sospecha de valvas de uretra posterior y obstrucción baja del tracto urinario. En forma diferida (después de 3 meses de vida) se puede solicitar frente a hallazgos ecográficos que hagan sospechas RVU.
Cintigrama dinámica: Indicado en sospecha de obstrucción de la vía urinaria. Mediante la inyección de un radioisótopo se evalúa la función diferencial y el drenaje renal. Es importante señalar que este estudio debe diferirse las primeras semanas de vida, dado que la inmadurez renal del recién nacido puede dificultar la interpretación del examen.
Resonancia magnética: De uso excepcional ya que requiere anestesia del paciente y es de alto costo. Es especialmente útil en el manejo de riñones obstructivos que tienen alteraciones anatómicas asociadas como riñón en herradura o ectopia renal cruzada.
Manejo postnatal
Uro profilaxis: cefadroxilo 10-15 mg/kilo, debe ser iniciado en todo paciente con HN severa o DAP >10mm prenatal en el tercer trimestre hasta que se descarte uropatía obstructiva. En el paciente que se confirma malformación debe mantenerse la uroprofilaxis hasta su posterior estudio y eventual corrección de la patología.
Circuncisión: se sugiere en pacientes con HN severa, RVU sintomático, síndrome Prunne Belly y VUP severa para disminuir el riesgo de ITU.
Obstrucción pieloureteral
La obstrucción pielo ureteral (OPU) es una patología caracterizada por obstrucción parcial del flujo de orina desde la pelvis renal hacia el uréter, generando hidronefrosis (HN) secundaria. Las causas de OPU pueden ser primarias – congénitas y secundarias, como, por ejemplo, vasos polares aberrantes. Su incidencia es de 1 de cada 500 recién nacidos vivos con HN prenatal y es la causa más frecuente de HN antenatal patológica(1,6). Afecta más a pacientes de genero masculino y a unidades renales izquierdas.
Clínica y Diagnóstico
La gran mayoría de las OPU son asintomáticas y se diagnostican mediante ultrasonografía prenatal. Cuando no hay seguimiento prenatal los recién nacidos pueden debutar con infección urinaria, masa abdominal, hematuria y déficit en el desarrollo pondoestatural. La presentación clínica en niños mayores puede ser dolor abdominal y/o dolor intermitente del flanco abdominal, daño renal ante traumatismos de bajo grado, hematuria, litiasis y/o hipertensión. En algunos casos la HN puede ser un hallazgo incidental durante el estudio ecográfico por otra causa como dolor abdominal.
El estudio inicial es con ecografia renal y vesical donde es fundamental evaluar grado de dilatación de la pelvis renal, presencia de dilatación calicilar, diferenciación corticomedular y grosor del parénquima renal. El diagnóstico se confirma mediante un cintigrama renal dinámico. Este examen entrega información de la función renal relativa e información del drenaje de orina desde el riñón hasta la fase post miccional.
Manejo
En general el manejo dependerá principalmente de cuan dilatado este la pelvis renal y de la función de esa unidad renal. La indicación quirúrgica será evaluada de acuerdo con los hallazgos ecográficos, la presentación clínica y el resultado de la cintigrama renal dinámico.
Indicaciones quirúrgicas en OPU:
- Patrón obstructivo en la cintigrama renal
- Función renal < 40%
- HN severa con DAP mayor a 30mm
- Aumento progresivo de la HN
- Adelgazamiento del parénquima renal con dilatación calicilar
- Pacientes sintomáticos (ITU, dolor abdominal, etc).
Fig. 6. Manejo de hidronefrosis prenatal
La técnica “Gold standard” es la pieloplastía de Anderson Hynes, que se puede realizar en forma abierta, laparoscópica/robótica. En algunos centros se están realizado endopielotomías con resultados a largo plazo aún en evaluación. La figura 6 resume los pasos a seguir en pacientes con diagnóstico antenatal.
Megauréter
Se considera como megauréter aquel que mide sobre 7mm de ancho en una ecografia, con o sin dilatación de la pelvis renal. Es resultado de una anormalidad funcional o anatómica a nivel de la unión uretero vesical. La principal causa corresponde a un segmento ureteral distal aperistáltico con una inserción en la vejiga normotópica. Existe también el megauréter secundario el cual es resultado de anomalías de la vejiga o de la uretra como vejiga neurogénica, valvas de uretra posterior, compresión tumoral entre otros. El megauréter primario es la segunda causa más frecuente en pacientes con diagnóstico prenatal de hidronefrosis, después de la obstrucción pieloureteral. Su incidencia se estima en 0,36 por 1000 recién nacidos vivos. Afecta con mayor frecuencia a hombres y al uréter izquierdo 1.
Clasificación
Los megaureteres primarios se clasifican según la presencia o ausencia de reflujo vesico ureteral y de obstrucción.
Megauréter Refluyente: Son aquellos uréteres que tienen asociado reflujo vesicoureteral habitualmente es de alto grado (IV – V).
Megauréter Obstructivo: Corresponde a aquel megauréter en el que se ha comprobado una obstrucción al flujo urinario a nivel de la unión ureterovesical. Sus principales causas son un segmento adinámico el uréter distal o una estenosis intrínseca del segmento distal de uréter. Este tipo de megauréter puede causar importante deterioro de la función renal por lo cual es importante su diagnóstico precoz y manejo.
Megauréter Obstructivo – Refluyente: Es el menos frecuente de todos los megaureteres primarios. Sin duda este grupo representa el mayor desafío diagnóstico. Menos del 5% de los pacientes presentan esta forma clínica en la que coexiste el reflujo vésico ureteral y la obstrucción.
Megauréter No Obstructivo – No Refluyente: La mayoría de los megaureteres caen en esta categoría. En estos pacientes existe una dilatación significativa del uréter, pero no se puede demostrar ni reflujo ni obstrucción subyacente. La mayoría de ellos son diagnosticados antenatalmente y muchos de ellos, entre 50 y 70%, son asintomáticos, pudiendo evolucionar a la resolución espontanea en el período post natal durante los dos primeros años de vida.
Clínica y Diagnóstico
Hoy en día el diagnóstico del megauréter es mayoritariamente en pacientes asintomáticos en una ecografía antenatal. Todo uréter diagnosticado prenatalmente debe ser estudiado en el periodo postnatal mediante ecografía renal y vesical (después del 3er día de vida), donde aquellos con diámetro mayor o igual a 7mm se consideran megaureteres. Si no hay diagnóstico antenatal estos pacientes suelen ser asintomáticos hasta que debutan con una infección urinaria, hematuria, dolor abdominal e incluso masa abdominal. El megauréter obstructivo suele ser el más sintomático ya sea con infección o dolor cólico. Ante una ecografía renal y vesical sugerente, el estudio debe completarse con una uretrocistografía miccional para diagnosticar reflujo vesicoureteral concomitante y para descartar obstrucción uretral (VUP). También se puede realizar cintigrama renal dinámico (MAG3) tanto para orientar en términos de función relativa renal y así como el diagnóstico de una obstrucción a nivel ureterovesical. Este último se recomienda realizarlo después de las primeras 4 semanas de vida para que tenga mayor validez.
Manejo
La mayoría de los pacientes con diagnóstico de megaureter caen en la categoría de no obstructivo, no refluyente, por esto la conducta en estos pacientes debe ser expectante, con control ecográfico seriado para monitorización de la dilatación renal y antibiótico profilaxis. Las indicaciones quirúrgicas no difieren mucho de las planteadas para una OPU, donde lo importante es el drenaje, la función renal, dilatación de cálices, calidad del parénquima y presencia o no de síntomas. Ante indicación quirúrgica en menor de 1 año, existen alternativas temporales para solucionar la obstrucción ya que intentar un neoimplante en una vejiga muy pequeña tiene riesgo de daño y denervación vesical. Se pueden realizar las siguientes opciones: una ureterostomía cutánea, un reimplante refluyente (ureterostomia interna), instalación endoscópica de un catéter doble J o una dilatación neumática endoureteral. En niños mayores el manejo quirúrgico que corresponde es el neoimplante vesicoureteral que corresponde al tratamiento definitivo.
Doble sistema pieloureteral, ureterocele y ureter ectopico
El doble sistema pieloureteral –DSPU– puede definirse como una unidad renal cuyo sistema colector esta conformado por dos sistemas pielocalicilares que drenarán en uno o dos uréteres. Puede ser unilateral o bilateral y pueden ser sintomáticos o no según las anomalías a las cuales se asocien.
El ureterocele corresponde a una dilatación quística de la porción submucosa intravesical del uréter generalmente asociado a un orificio ureteral estenótico. Su etiología no ha sido aclarada. Puede asociarse a un DSPU o bien, presentarse en un sistema único, lo que en pediatría es poco frecuente. En los DSPU, el ureterocele se relaciona con el uréter del polo superior y con frecuencia causa obstrucción y daño renal secundario a ese polo renal (1,6).
El uréter ectópico es aquel que desemboca caudal a la inserción normal en el trígono vesical. Sobre el 75% de los uréteres ectópicos se asocian a un DSPU y corresponden al uréter del sistema superior. Tanto en sistemas únicos o dobles, la ectopia ureteral puede asociarse a una unidad renal hipoplasica e hipofuncionante.
Por otro lado, en los DSPU existe RVU asociado con el uréter del polo inferior y se debe a una inserción anómala alta.
Incidencia
El DSPU ocurre en el 0,8% de la población general siendo la gran mayoría de estos pacientes asintomáticos. Su presentación según género es 2 veces mas frecuente en mujeres y según lateralidad, es semejante. Hasta un 20% de los casos se presenta de manera bilateral y solo el 30% corresponde a un DSPU completo.
Ureterocele: Incidencia reportada 1 en 4 mil niños, siendo 4 a 7 veces más frecuente en mujeres. El 80% se asocia al sistema superior de un DSPU y 20% en sistemas únicos. 10% de los ureteroceles son bilaterales. Se clasifican según su ubicación en ectópico e intravesical u ortotópico.
Ureterocele ectópico: Se refiere a la situación en la cual parte del ureterocele se extiende hacia el cuello vesical o uretra. Es la forma más frecuente de ureterocele en DSPC (Doble Sistema Pieloureteral Completo) alcanzando el 80% de los casos. Dependiendo del tamaño, estos ureteroceles pueden generar problemas a los otros uréteres como obstrucción al uréter del polo inferior ipsilateral o al uréter contralateral. En otras ocasiones puede levantar el piso vesical causando reflujo del uréter del sistema inferior ipsilateral. Cuando el ureterocele prolapsa a través de la uretra lo denominamos un ceco-ureterocele. Este último puede causar obstrucción uretral y repercusión en ambas unidades renales.
Intravesical: el ureterocele se encuentra en su totalidad dentro de la vejiga. Corresponden al 15% de los casos y con frecuencia se asocian a sistema renal único.
Uréter ectópico: su incidencia es difícil de establecer puesto que hay un porcentaje de pacientes asintomáticos sin diagnostico. Se estima que esta es alrededor de 1 por cada 1900 niños. Es 6 veces más frecuente en pacientes del género femenino. Existen diferencias de genero en cuanto a la desembocadura de uréter ectópico y, por ende, de la sintomatología que los caracteriza. En niños, el 50% desemboca en uretra posterior. Otros sitios posibles son las vesículas seminales, conducto deferente, conductos eyaculadores, etc. Pero siempre lo hacen proximal al esfínter urinario externo de tal manera que estos pacientes no presentan incontinencia urinaria secundaria al uréter ectópico. En las niñas, la desembocadura casi siempre es distal al esfínter urinario externo y la sintomatología clásica es la incontinencia urinaria. Los principales sitios de llegada del uréter ectópico en niñas es cuello vesical, uretra proximal, vagina, vestíbulo vaginal entre otros.
Clínica y Diagnóstico
Un DSPU incompleto, sin dilatación asociada, es frecuente y suele ser asintomático y su diagnóstico se efectúa por un hallazgo ecográfico. Generalmente no requiere estudio complementario ni tratamiento. En DSPC, los problemas tienen relación con la llegada de los uréteres a la vejiga. El uréter del polo superior puede insertarse normalmente en la vejiga, puede ser ectópico o asociarse a un ureterocele. El uréter polar inferior puede insertarse normalmente o bien, insertarse lateral al trígono con lo cual su trayecto intramural resulta más corto y se asocia a reflujo vesico ureteral. Estos últimos suelen presentarse clínicamente con ITU y, en el estudio correspondiente, se identifica en la ecografía una hidroureteronefrosis leve a moderada polar inferior y en la uretrocistografía (UCG) se confirma el RVU.
El diagnóstico de ureterocele puede ser prenatal o postnatal. En ecografías prenatales puede encontrarse una hidroureteronefrosis (HUN) y menos frecuentemente se observa una imagen de ureterocele intravesical. En el periodo postnatal, estas malformaciones habitualmente se presentan con una infección urinaria en los primeros meses de vida. El diagnostico de ureterocele se realiza mediante ecografía renal y vesical y UCG. En la ecografía puede observarse la imagen quística intravesical asociada a HUN del sistema comprometido. La UCG nos debiese confirmar la presencia de ureterocele durante el llene vesical al verse imagen de masa intravesical que no capta el medio de contraste. La UCG también es relevante para determinar si hay reflujo presente en el sistema polar inferior o del sistema contralateral. El estudio debe completarse con un cintigrama renal funcional, de tal manera de determinar función renal diferencial y establecer obstrucción de vaciado en el polo superior comprometido.
En relación con los uréteres ectópicos asociados a DSPU, también podemos establecer diagnóstico prenatal y postnatal. En ecografías prenatales suele encontrarse hidronefrosis con uréter visible. Postnatal, estos pacientes suelen presentar ITU en cuyo estudio ecográfico se identifica HUN del sistema con uréter ectópico. En niñas mayores la clínica habitual es la incontinencia urinaria y en niños mayores aparecen síntomas como dolor abdominal, urgencia miccional (uréter desemboca en uretra posterior), epididimitis (uréter desemboca en conducto deferente). El estudio diagnóstico se inicia con una ecografía renal y vesical que frecuentemente demuestra HUN con uréter dilatado hacia distal en una posición anormalmente baja. Cuando además hay compromiso funcional renal, se ve un riñón o un polo renal superior displástico y pequeño.
Para identificar la llegada real del uréter el estudio puede complementarse con una uretrocistografía que demuestre reflujo a este uréter. Cuando no hay claridad diagnóstica, la siguiente etapa es realizar una uroresonancia con contraste que confirma el diagnóstico y muestra la desembocadura del uréter. El estudio debe completarse con un cintigrama renal de tal manera de determinar la función renal o la función del polo superior en caso de ser un DSPU. La función del polo o unidad renal comprometido tiene relevancia al tomar decisiones quirúrgicas.
Tratamiento
A excepción de DSPU sin patología asociada, muchas de las otras malformaciones urológicas descritas requerirán de manejo quirúrgico. En presencia de un DSPU con RVU del sistema inferior, el manejo del reflujo es semejante al de reflujo en sistema único. En pacientes con reflujo asociado, se sugiere mientras se realiza estudio y se decide conducta, mantener uroprofilaxis.
Al enfrentar un ureterocele tenemos varios elementos que considerar al momento de planificar la resolución quirúrgica:
- Condición clínica del paciente (ej. hallazgo vs. uro sepsis)
- Tipo de ureterocele (ectópico / intravesical)
- DSPU o sistema único
- Función renal del polo afectado
- Presencia de RVU
- Presencia de obstrucción ya sea ipsilateral, contralateral o de la uretra
Tras considerar todos estos elementos se debe optar por la alternativa de tratamiento para cada paciente en particular. Estas incluyen la observación, la punción endoscópica del ureterocele, la heminefroureterectomia polar superior, reparación de piso vesical con eventual reimplante y la uretero-uretero anastomosis.
El tratamiento del uréter ectópico asociado a DSPU también puede ser quirúrgico. En HUN leve-moderada asintomáticas se puede observar. En casos de DSPU con polo superior no funcionante, el tratamiento puede ser a heminefroureterectomia. Cuando el polo superior tiene función conservada, el tratamiento es la piel uretero anastomosis alta o la uretero-uretero anastomosis baja.
Valvas de uretra posterior
La mayoría de las obstrucciones uretrales en pediatría son congénitas y de todas las causas de obstrucción uretral, las valvas de uretra posterior (VUP) son las más frecuentes. Su relevancia esta dada porque causan cambios secundarios graves en el tracto urinario superior, que conducen a insuficiencia renal crónica terminal en más del 50% de los pacientes diagnosticados con esta patología. Las VUP son membranas obstructivas dentro del lumen de la uretra que se extienden desde el verumontanum hacia distal. Sólo se presentan en hombres, y es una de las principales causas de daño renal y trasplante en la población pediátrica. La incidencia ha sido estimada aproximadamente en 1 por 5000 a 8000 recién nacidos vivos masculinos. Mientras más temprano se presenta la obstrucción durante la embriogénesis, mayor probabilidad de daño renal (1,7).
Clínica y diagnóstico
Presentación prenatal: Se puede diagnosticar desde la semana 8va de gestación, pero generalmente se hace entre las semanas 16 y 20. Cerca de la mitad de estos casos se diagnostican después del segundo trimestre, en estudios de rutina. Se puede manifestar con oligohidroamnios, hipoplasia pulmonar, ascitis urinaria, urinomas perinéfricos; y en la ecografía hidroureteronefrosis uni o bilateral, vejiga distendida (megavejiga) con paredes engrosadas. Generalmente, los pacientes diagnosticados antes de las 16 semanas tienen un mal pronóstico, sobre todo si están asociados a oligohidroamnios, lo que puede llevar a hipoplasia pulmonar y muerte fetal (síndrome de Potter).
Presentación postnatal: Las formas de presentación en periodo neonatal incluyen: insuficiencia renal terminal precoz, vejiga palpable, masa abdominal palpable (hidronefrosis o urinoma), ascitis urinaria, chorro miccional disminuido, infección urinaria, rechazo alimentario y mal incremento ponderal. En lactantes, generalmente la presentación es con infección urinaria y en ocasiones asociada a insuficiencia renal aguda, aunque también se pueden presentar con falla en el crecimiento pondoestatural e insuficiencia renal crónica. En escolares la presentación es similar a los lactantes, sin embargo, se puede sospechar con cuadros de disfunción miccional con una micción prolongada y chorro miccional disminuido.
Estudios complementarios
Es imperativo evaluar la función renal y la depuración de creatinina para determinar el manejo a seguir de estos pacientes, así como para estabilizar en el momento agudo. Se deben solicitar hemograma, electrolitos séricos, creatinina, nitrógeno ureico, calcio, fósforo y gasometría arterial o venosa. La evaluación de la creatinina en el recién nacido debe contemplar que hasta los 3-5 días de vida, la creatinina fetal corresponderá a la creatinina materna, por lo que es recomendable hacer una curva de creatinina.
Ecotomografía: Se utiliza en el diagnóstico prenatal y postnatal, valorando la presencia de dilatación de uretra posterior, paredes vesicales, presencia de uréter distal, hidro o hidroureteronefrosis y parénquima renal.
Uretrocistografía miccional: Provee información definitiva del diagnóstico con la imagen de dilatación de uretra posterior. Además, evidencia la presencia de RVU (40- 60% de los casos). Cistoscopia: Establece el diagnóstico definitivo ya que se observan directamente las VUP, además de poder realizar el tratamiento en el mismo acto quirúrgico. Se utiliza también para determinar el estado de la vejiga y de los meatos ureterales.
Urodinamia/videourodinamia: Es necesaria para evaluar la función de la vejiga, tanto inicialmente como en el seguimiento de los pacientes a corto y largo plazo. Incluso para determinar cambios en el manejo médico y/o quirúrgico.
Manejo
Fetal: Teóricamente, la liberación de la obstrucción in útero podría disminuir el daño renal colocando un shunt vésico-amniótico. Sin embargo, el pronóstico renal de estos pacientes sigue siendo muy malo y la colocación del shunt sólo ha demostrado mejorar la sobrevida fetal, pero sin tener resultados consistentes en el pronóstico renal.
Recién nacidos o lactantes: Lo primordial es controlar la insuficiencia renal y las alteraciones electrolíticas. Para mejorar la insuficiencia renal aguda es necesario liberar la ![]() obstrucción, lo cual se puede obtener con una vesicostomía, ablación de valvas uretrales, ureterostomía (unilateral o bilateral) o pielostomía (unilateral o bilateral).
obstrucción, lo cual se puede obtener con una vesicostomía, ablación de valvas uretrales, ureterostomía (unilateral o bilateral) o pielostomía (unilateral o bilateral).
Preescolares-escolares: El tratamiento a largo plazo depende de la evolución de cada paciente, de su función renal y del estado de su vejiga. Es decir, si presenta disfunción miccional, en ocasiones puede ser manejada con kinesioterapia, pero otros pacientes van a requerir de cateterismo limpio intermitente (CLI), para lo cual sería necesario realizar una derivación urinaria continente tipo Mitrofanoff, ya que los pacientes con VUP tiene sensibilidad uretral. En muy pocos casos el aumento vesical, con uréter o con segmento intestinal (enterocistoplastía) es necesario, pero debe tenerse en consideración en aquellos pacientes con mala compliance vesical y/o hiperactividad refractaria al tratamiento médico.
Complicaciones y Pronóstico1: En general se pueden resumir en disfunción miccional, infección urinaria a repetición, reflujo vesicoureteral, y daño renal crónico, siendo este el más grave de todos y pudiendo la mayoría requerir de trasplante renal en el futuro. El pronóstico a largo plazo de los pacientes con VUP está determinado por una combinación del grado de desarrollo anormal, incluyendo la uretra, la vejiga, los riñones y los uréteres; y la gravedad de los efectos secundarios de la obstrucción del tracto de salida de la vejiga y el tracto urinario superior. Cerca de un 50% de los pacientes con VUP tendrán daño renal crónico terminal, y los factores que influyen en esto son:
- Displasia renal primaria.
- Daño renal secundario como resultado de obstrucción al tracto de salida de la vía urinaria.
- Infección urinaria, asociado a RVU.
- Disfunción miccional. Típicamente, la hiperactividad del detrusor será reemplazada por falla del mismo, acompañada de disminución de la capacidad vesical e incremento de los volúmenes residuales. Esto puede condicionar a una disfunción miccional grave, lo cual puede agravar el daño renal previamente establecido. El seguimiento a largo plazo de los pacientes con VUP requiere de un rastreo estrecho, tanto de la función renal como del comportamiento vesical, para mejorar o corroborar la disfunción miccional.
Sindrome de Prune Belly
También conocido con Síndrome de Abdomen en Ciruela-Pasa el Síndrome de Prune Belly se caracteriza por se una asociación caracterizada por la triada de: agenesia, hipoplasia o deficiencia de la musculatura de la pared abdominal; hidronefrosis en distintos grados y criptorquidia bilateral. La incidencia estimada es de 1 en 30.000 – 40.000 RN, correspondiendo a pacientes del sexo masculino en un 95%. Las manifestaciones varían entre los distintos individuos, siendo un factor pronóstico el compromiso renal7.
Etiología
La etiología de este síndrome no está del todo clara. Existen teorías que podrían explicar el origen de éste, pero ninguna es universalmente aceptada.
- Obstrucción de la uretra posterior en etapas tempranas del desarrollo embrionario, produciendo dilatación del tracto urinario, que condicionaría el defecto en la pared abdominal e impediría el descenso testicular
- Defecto primario del mesodermo, que condicionaría las alteraciones del tracto urinario y de la pared abdominal.
- Defecto intrínseco del tracto urinario, produciendo dilatación ureteral y ascitis fetal.
Clasificación
Se clasifica en tres grupos, dependiendo de la severidad de las manifestaciones clínicas, lo que condiciona el pronóstico:
Categoría I: Anormalidades urinarias severas, con compromiso pulmonar importante, con elevada morbimortalidad en el periodo neonatal secundaria a insuficiencia renal o respiratoria.
Categoría II: Compromiso de las vías urinarias moderado, sin trastornos respiratorios asociados, con el tratamiento quirúrgico mejora la función renal lo que permite una sobrevida cercana al 80%.
Categoría III: Manifestaciones leves, sin compromiso de la función renal, mortalidad baja.
Clínica
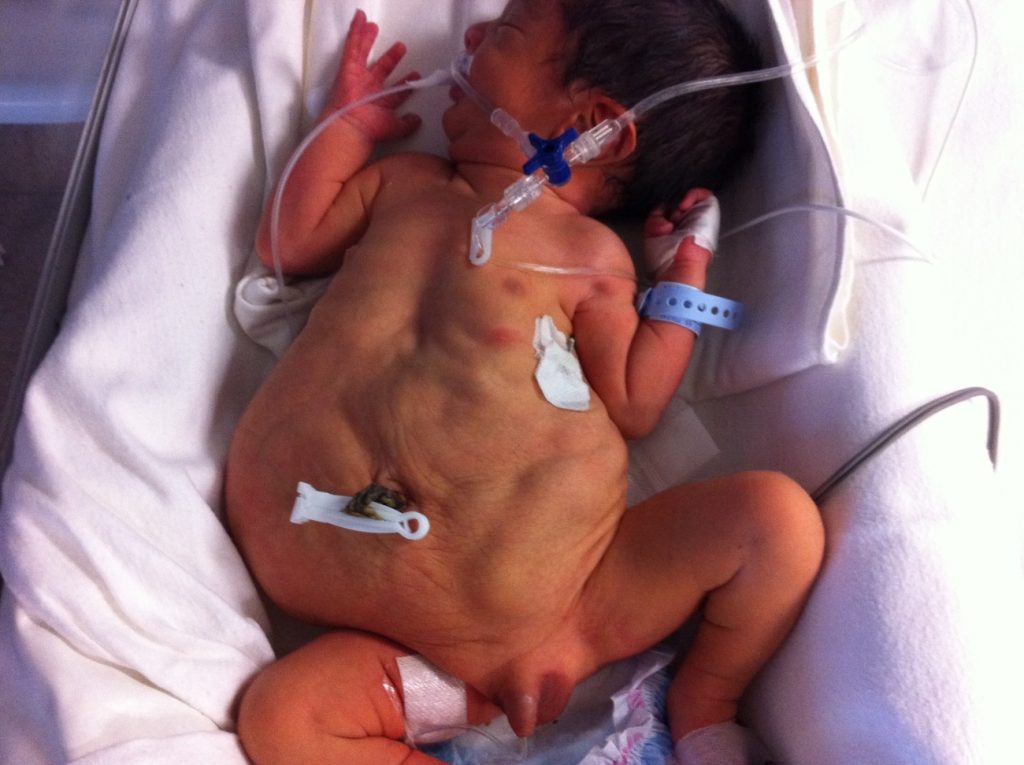
Manifestaciones genito urinarias: La displasia renal es la alteración más frecuente y generalmente es asimétrica. El compromiso renal es el principal factor pronóstico en estos pacientes. Se observa algún tipo de compromiso ureteral en todos los casos, especialmente hacia distal, secundario a hipoplasia de células de musculo liso, lo que se traduce en uréteres aperistálticos, dilatados y tortuosos. Existe asociación con RVU en un 75-85% de los casos. Se observa megavejiga con pseudodivertículo o persistencia del uraco, de paredes engrosadas a expensas de tejido conectivo, con un trígono grande, asimétrico y orificios ureterales en posición lateral. La compliance vesical es adecuada, la contractibilidad se ve comprometida en la mitad de los casos, lo que se ve reflejado en residuos postmiccionales elevados (figura 7). La uretra posterior se encuentra dilatada lo que podría explicarse por posible atresia, estenosis o válvulas hacia distal, lo que determina una próstata hipoplásica. La criptorquidea es uno de los pilares diagnósticos del síndrome, lo más común es que se encuentren intraabdominales, a nivel de los vasos iliacos. La fertilidad esta potencialmente disminuida, pese a que el estudio histológico es normal, por alteraciones en la maduración de las células germinales y por factores extratesticulares como disyunción epididimaria, pero eso no necesariamente implica que la paternidad este afectada.

Manifestaciones extra genitourinarias: Además de las alteraciones de la pared abdominal, el 75% de los pacientes presenta otras anomalías asociadas:
Defecto de la pared abdominal: Lo más característico es el compromiso de la musculatura inferomedial, lo que produce el aspecto característico con la piel del abdomen arrugada y redundante que cae hacia los costados. En partes del cuerpo más afectadas solo se observa piel, celular y aponeurosis. (figura 8)
Cardiopatías: Presentes en el 10% de los pacientes, se ha observado relación con Ductus Arterioso Persistente, CIA, CIV y Tetralogía de Fallot.
Anomalías Pulmonares: Se observa hipoplasia pulmonar en pacientes con antecedentes de oligohidroamnios, a lo que se puede asociar pneumotórax o neuromediastino. La mecánica de la tos se ve interferida por el compromiso abdominal, lo que se asocia a mayor incidencia de neumonías y atelectasias.
Alteraciones del tracto gastrointestinal: Se puede encontrar malrotación intestinal hasta en un 30% de los casos. Otras alteraciones observadas son ano imperforado, atresias y estenosis intestinales, entre otros.
Alteraciones esqueléticas: Observadas hasta en un 45% de los casos, en su mayoría son secundarias al oligohidroamnios como pie equino, escoliosis y displasia congénita de caderas.
Diagnóstico
Periodo Antenatal: El hallazgo en el estudio ecográfico prenatal precoz (11 a 14 semanas de gestación) de hidroureteronefrosis es inespecífico. La observación de megavejiga, dilatación ureteral bilateral y distención abdominal irregular a las 30 semanas es altamente sugerente del Sindrome. de Prune Belly.
Periodo Postnatal: Al nacer si no hay sospecha antenatal, el aspecto característico del abdomen en asociación con testículos no palpables es suficiente para el diagnóstico.
Estudio Complementario
Una vez establecido el diagnóstico y estabilizado el paciente se debe estudiar con:
-
- Radiografía de Tórax: para determinar grado de hipoplasia pulmonar.
- Ecocardiográma: evalúa asociación con cardiopatías.
- Estudio de Vía Urinaria: inicialmente se realiza Ecotomografía renal y Vesical, posteriormente se complementa con Uretrocistografía y Cintigrama Renal.
- Estudio de Función Renal: creatinina, nitrógeno ureico, gases y electrolitos.
Tratamiento
El manejo es multidisciplinario, incluyendo a pediatras, nefrólogos y urólogos. Inicialmente se debe compensar la parte respiratoria y determinar el grupo al que corresponde el paciente y su pronóstico. En la Categoría I, dado que la mortalidad es prácticamente del 100% y solo estarían indicados cuidados de soporte básico. En los demás pacientes una vez estabilizados y compensados desde el punto de vista renal, se deberá analizar caso a caso si es necesario y como realizar la reconstrucción de la vía urinaria. En casos severos se recomienda una derivación urinaria precoz y diferir la reconstrucción de la vía urinaria una vez compensado el paciente. El descenso testicular se recomienda a los 6 meses de vida, preferentemente por vía intrabdominal, para obtener una adecuada movilización de los vasos espermáticos.
La abdominoplastía además de un rol cosmético, mejoraría la dinámica pulmonar, digestiva y vesical. Existen variadas técnicas, cuyo fin es resección de la piel redundante y plicatura de la fascia. Idealmente la reconstrucción de la vía urinaria, orquidopexia y abdominoplastía podrían ser realizadas en un tiempo quirúrgico. Esta decisión dependerá del estado previo del paciente y de la experiencia del equipo quirúrgico7.
Incontinencia urinaria y enuresis
La incontinencia urinaria diurna y/o nocturna (enuresis) pediátrica es una entidad clínica muy frecuente, que motiva una demanda importante de la consulta urológica pediátrica. Se estima que entre un 10-15% de los niños mayores de 5 años presentan incontinencia urinaria diurna y/o nocturna8. Pese a ser un problema muy frecuente, suele ser diagnosticado y tratado de manera inadecuada, generando frustración en los pacientes y sus familiares.
El “niño que se moja” en muchas ocasiones representa un problema de carácter benigno que se resolverá en forma espontánea junto con el crecimiento y maduración del paciente. Sin embargo, en ocasiones puede ser manifestación de una enfermedad urológica subyacente y/o implicar un detrimento social y psicológico importante para el paciente y su familia. Las manifestaciones clínicas son variadas y deben ser exploradas acuciosamente en la historia clínica. La terminología con la que se ha investigado y tratado este tema muchas veces ha sido equívoca, confusa y utilizada en forma indistinta a la incontinencia del adulto. Por esta razón, en el año 2006, la sociedad internacional de continencia infantil ha propuesto una estandarización, la que resulta fundamental para un adecuado diagnóstico y tratamiento9. La figura 9 resume los principales elementos para clasificar la incontinencia urinaria en niños.

Historia clínica
- Hábito miccional/intestinal: Frecuencia diaria reportada, ingesta de líquidos (tipos), inicio del control de esfínter, consistencia de deposiciones (recomendamos utilizar escala de Bristol), alimentación en general. Se interrogará en forma dirigida por síntomas urinarios asociados tales como:
- Urgencia miccional
- Latencia miccional
- Dificultad para vaciamiento vesical
- Chorro débil
- Polaquiuria
- Tenesmo
- Presencia o no de incontinencia nocturna (enuresis)
- Maniobras de retención urinaria/fecal (Cruzar piernas, movimientos de extremidades, etc)
- Goteo post miccional
- Historia familiar de incontinencia urinaria nocturna (enuresis). Calidad y hábitos del sueño
- Concomitancia con infecciones del tracto urinario altas o bajas y/o otra patología urológica
Examen Físico
Pese a que la mayor parte de los niños que consultan por incontinencia urinaria son sanos, es importante no olvidar realizar un examen físico completo, determinando el estado general y nutricional del paciente. En forma dirigida se explorará el abdomen en busca de masas (hidronefrosis/tumores) y/o vejiga palpable (vejiga neurogénica). El examen genital debe consignar la ubicación del meato, la existencia de malformaciones anatómicas evidentes (epispadias, seno urogenital, uréter ectópico), la apariencia del introito vaginal en niñas, descartando la presencia de sinequias de labios menores y consignado la presencia/no de eritema vulvar. Se debe explorar la región lumbosacra en búsqueda de estigmas de alguna disrrafia medular oculta (hemangiomas, manchas pilosas, fosita pilonidal).
Estudio básico
- Cartilla miccional/defecatoria: Elemento fundamental en la evaluación de la incontinencia urinaria en el niño. La tabla 2 enumera los elementos a considerar al elaborar un registro miccional/defecatorio.
- Ecografía renal y vesical pre-post miccional: Se solicita en todos los pacientes, permite evaluar la presencia de malformaciones renales (doble sistema con uréter ectópico, hidro ureteronefrosis) y/o vesicales como engrosamiento vesical, residuo post miccional aumentado y/o signos sugerentes de vejiga neurogénica.
- Uroflujometría con electromiografía de superficie: Estudio no invasivo que permite evaluar el flujo urinario máximo, la curva flujo/volumen y la actividad electromiográfica durante la micción. En la figura 10 se representan los tipos de curva flujo/volumen normal y en “stacatto” característica de la incoordinación detrusor-esfinteriana.
| DATOS | PERIODO | INFORMACION |
| Hora y volumen (ml) urinario | Mínimo 48 horas | Frecuencia miccional, volumen máximo, mínimo y promedio |
| Episodios de incontinencia | 14 días | |
| Episodios de Enuresis | 14 días | |
| Volumen Enuresis (peso pañal) | 7 días | Presencia poliuria nocturna |
| Hábito intestinal | 14 días | Asociación con constipación |
| Hora de acostarse/levantarse | 14 días | Horas totales de sueño |
Tabla 3. Cartilla miccional/defecatoria (modificado de Neveus et al, 2006)
Fig. 10. Curvas de uroflujometría
Estudio complementario avanzado
De acuerdo con el estudio inicial y si existe la sospecha de patología urológica compleja, se solicitarán estudios complementarios avanzados, tales como uretrocistografía miccional, urodinamia y/o videourodinamia, resonancia de columna lumbar, entre otros. Los exámenes invasivos deben racionalizarse y solicitarse exclusivamente frente a cuadros refractarios al tratamiento habitual y/o en pacientes con compromiso de la función renal.
Diagnóstico
Una vez efectuada una correcta anamnesis, examen físico y evaluando el resultado del estudio complementario, el niño que se moja podrá ser catalogado en un diagnóstico presuntivo más exacto que permitirá adecuar de mejor forma su tratamiento
Enuresis
Se define como enuresis a la pérdida de orina involuntaria durante el sueño mayor o igual a tres veces a la semana, en un niño mayor de 5 años. Tiene una prevalencia aproximada del 5% y es más frecuente en el sexo masculino. Esta condición tiene una asociación familiar y se estima que, si ambos padres tienen antecedente de enuresis, la posibilidad de tener un hijo con esta condición es cercana al 80% (8-9). La patogenia de esta condición no esta completamente establecida, sin embargo, se ha establecido que hasta la mitad de los pacientes enuréticos tienen poliuria nocturna debido a una falla en el ritmo circadiano de la producción de hormona antidiurética. Otro grupo de niños pueden presentar hiper actividad del detrusor durante el sueño o tienen alterado el mecanismo del “despertar”. La enuresis a su vez se clasifica en:
- Primaria monosintomática: Paciente mayor de 5 años que no ha logrado continencia nocturna o del sueño por período mayor a 6 meses. A la exploración dirigida, no hay ninguna evidencia de sintomatología de disfunción del tracto urinario (urgencia, retención, disuria, tenesmo, polaquiuria, volumen urinario bajo) y/o incontinencia diurna.
- Primaria no monosintomática o mixta: Generalmente los familiares que consultan por enuresis tienden a subestimar la sintomatología diurna, la que muchas veces es compensada por el paciente. Sin embargo, la enuresis mixta es la más frecuente. Se presenta en pacientes mayores de 5 años que no ha logrado continencia nocturna o del sueño por período mayor a 6 meses. A la exploración dirigida, hay evidencia de sintomatología de disfunción del tracto urinario (urgencia, retención, disuria, tenesmo, polaquiuria, volumen urinario bajo) y/o incontinencia diurna.
- Enuresis secundaria: Infrecuente, se diagnostica en pacientes mayores de 5 años que han pasado por un período mayor a 6 meses de continencia nocturna completa. Se deben descartar factores psicológicos y/o abuso infantil.
Incontinencia diurna (8-9)
- Vejiga Hiperactiva: El síntoma cardinal es la urgencia miccional y se asocia frecuentemente a incontinencia diurna de cuantía variable. Muchas veces se observa un aumento de la frecuencia miccional y la cartilla revela volúmenes urinarios pequeños.
- Retenedores/vejiga “floja”: Pacientes que tienen la frecuencia miccional disminuida, asociado/no a maniobras de retención urinaria. Generalmente tienen la capacidad vesical aumentada para la edad y se puede asociar a residuo post miccional elevado. Frecuentemente se asocia a distintos grados de constipación.
- Disfunción miccional / Disinergia detrusor esfinteriana: Paciente que presenta contracción esfinteriana durante la micción. Pueden o no presentar síntomas de hiperactividad y/o retención. La curva de la uroflujometría revela un patrón en stacatto. La ecografía puede mostrar residuo post miccional elevado.
- Incontinencia de esfuerzo: Extremadamente rara en niños, frecuentemente se trata de pacientes con vejiga hiperactiva y/o pacientes con incontinencia durante la risa.
- Reflujo Uretrovaginal: Pacientes que presentan característicamente goteo post miccional y/o incontinencia post micción. No hay síntomas de urgencia. El examen físico puede revelar un introito profundo en pacientes obesas o la presencia de sinequias de labios menores.
- Incontinencia de risa: Infrecuente, se produce sólo cuando el paciente se ríe y no existen síntomas urinarios en otros contextos. Debe ser diferenciada de la vejiga hiperactiva, que es mucho más frecuente y también puede manifestarse con incontinencia de este tipo.
Tratamiento (8-9)
- Uroterapia: Conjunto de medidas no farmacológicas en relación con los hábitos del paciente. Se debe insistir en la importancia en la ingesta de agua, el consumo de frutas y verduras y el aseo genital adecuado. Se instará a tener un patrón miccional/defecatorio conocido, sin postergar el deseo miccional, orinando cada 2-3 horas y evacuando deposiciones blandas todos los días.
- Kinesioterapia de piso pelviano: Indicada en pacientes mayores de 4 años que puedan colaborar con la terapia. Permite reforzar los hábitos de uroterapia, mejorar la localización de la musculatura del piso pelviano y optimizar el vaciamiento vesical. Puede ser indicada como monoterapia o como complemento a las terapias farmacológicas. La técnica de miobiofeedback permite a los niños la visualización de la actividad muscular del piso pelviano y está especialmente indicada en pacientes con disinergia detrusor esfinteriana.
- Alarmas Enuréticas: Es la terapia de elección en pacientes con enuresis monosintomática. Requiere alta motivación tanto del paciente como de su entorno familiar. Funciona con un sensor de humedad que activa una chicharra, lo que permite que el paciente despierte y orine en el baño. Su efectividad es cercana al 70% y tiene baja tasa de recidiva.
- Terapia Farmacológica:
- Anticolinérgicos: La terapia anticolinérgica es una de las formas de tratamiento más habitual para la vejiga hiperactiva. Su uso puede mejorar la capacidad vesical y mejorar la “compliance” en pacientes neurogénicos, así como disminuir la hiperactividad vesical en incontinencias no neurogénicas. En pediatría el anticolinérgico más utilizado es la oxibutinina en su presentación clásica (dosificada cada 8 horas) y en liberación prolongada. Hay que tener precaución sobre la aparición de efectos secundarios (sequedad bucal, constipación, rubor facial), los que son dosis dependientes. También es importante evaluar el vaciamiento vesical del paciente, pues puede empeorar al iniciarse la terapia con anticolinérgicos. Otros anticolinérgicos como Tolterodina y Solifenacina también han sido utilizado en niños, especialmente en aquellos con mala tolerancia a la Oxibutinina.
- Toxina Botulínica: La inyección de toxina botulinica intravesical está principalmente indicada en hiperactividad de origen neurogénico, pero también se ha utilizado en pacientes funcionales en los que han fracasado las terapias convencionales. Su uso en estos pacientes es excepcional y puede provocar deterioro del vaciamiento vesical que requiera cateterismo intermitente.
- Bloqueadores alfa-agonistas: En pacientes con disinergia detrusor esfinteriana severa, el tratamiento con bloqueadorea alfa puede ser una opción terapéutica. Está indicada en pacientes con vaciamiento vesical incompleto, que no responden a terapia kinésica o no han iniciado el control de esfínter.
- Desmopresina: Análogo de la hormona antidiurética, es altamente eficaz (60-80%) en pacientes con enuresis monosintomática. Especialmente indicado en aquellos que presentan poliuria nocturna. El mayor problema es que se trata de un tratamiento sintomático, por lo que presenta una recidiva cercana al 40% al descontinuar su uso.
Reflujo vesico ureteral e infección urinaria
Se define como reflujo vésico ureteral (RVU) al paso de orina desde la vejiga hacia el uréter y/o el sistema colector renal. Es una patología urológica frecuente, estimándose una incidencia de aproximadamente un 1-2% de los niños. Sin embargo, esta podría ser mayor, pues existe un número indeterminado de población pediátrica con RVU asintomático y, por ende, no diagnosticado. Posterior a un episodio de pielonefritis en la infancia, la incidencia aumenta hasta un 30% y se estima que cerca de un 10% de los pacientes con diagnóstico antenatal de hidronefrosis tendrán RVU como causa subyacente.
Clasificación y etiología
La clasificación clasifica del RVU lo tipifica en primario, cuando hay una alteración anatómica de la unión uretero vesical con pérdida del sistema valvular y en secundario, cuando existe una causa subyacente que genera aumento significativo de la presión intravesical que desencadena el reflujo, como es el caso de la vejiga neurogénica y las valvas de uretra posterior. Sin embargo, hoy en día se sabe que en el RVU primario, las alteraciones del vaciado vesical funcionales también condicionan aumento de presión al tracto de salida urinario y por ende juegan un rol importante en la perpetuación y falla en el tratamiento de esta patología.
La etiología del reflujo aún no está completamente comprendida. Se sabe que el RVU en sí mismo no provoca sintomatología ni ocasiona daño renal. Sin embargo, la presencia de reflujo facilita la colonización del tracto urinario alto por gérmenes que pueden estar presentes en el tracto urinario inferior, desencadenando una infección renal que podría provocar daño renal.
Es importante comprender que la alteración de la función renal presente en algunos pacientes con RVU o también llamada “nefropatía por reflujo”, es secundaria a dos factores: displasia renal y post infecciosa. Un porcentaje importante será por displasia renal, es decir son pacientes en los que existe una alteración congénita de la formación renal y, por ende, hipofunción renal desde el nacimiento. Otro grupo de niños tendrá deterioro de la función renal secundario a infección, generalmente por episodios reiterados de pielonefritis durante la infancia precoz. Existe un componente hereditario de esta patología y, si bien no está indicado en pacientes asintomáticos, se estima que los hermanos de los pacientes tienen RVU hasta en un 47%.
Historia clínica
La mayoría de los pacientes que presentan RVU son diagnosticados después de un episodio de infección del tracto urinario (ITU). La historia típica es la de un lactante en el que, luego de un episodio de pielonefritis, se diagnostica esta condición. Aproximadamente un 15% de los pacientes portadores de RVU son diagnosticados en el contexto del estudio de un diagnostico ante natal de hidronefrosis. Otras formas de presentación incluyen: estudio por insuficiencia renal, hallazgo ecográfico de hidroureter o asimetría renal sugerente de daño renal y estudio de hermanos portadores de reflujo (indicado sólo en infección urinaria). En la presentación sintomática, con infecciones urinarias, con frecuencia observamos dos escenarios clínicos. El primero, es el de un lactante varón menor de dos años, no circuncidado, con cuadro febril y estudio compatible con pielonefritis. La otra presentación es en preescolares mujeres, en las que coincidentemente con la edad de retiro de pañal (entre los 2-3 años), presentan uno o múltiples episodios de infección urinaria, algunos o no febriles. Estas pacientes con frecuencia tienen alteración de la función vesical e intestinal, también llamado bowel and bladder dysfunction (BBD) y el estudio es compatible con RVU. El pronóstico y enfrentamiento en ambos casos es completamente diferente por lo que el tratamiento se deberá ajustar a la condición clínica del paciente.
Estudio
- El estudio inicial de un paciente pediátrico que se presenta con una ITU comprende una ecografía renal y vesical. Este estudio tiene bajo rendimiento para el diagnóstico de RVU. Algunos hallazgos que podría sugerir la presencia de esta condición incluyen: asimetría renal y/o cicatrices renales y presencia de hidroureter de cuantía leve-moderada que fluctúa durante el examen (aumenta durante la micción). La presencia de una hidroureteronefrosis severa es poco sugerente de esta condición y obliga a descartar en primer lugar patología obstructiva.
- Urosonografía miccional: Estudio ecográfico de RVU mediante la administración de un contraste intravesical. El más utilizado es mediante la utilización de microburbujas de hexafluoruro sufúrico (SonoVue®). Tiene una sensibilidad entre 60-100% para el diagnóstico con una especificidad cercana al 90%.
- Cintigrama renal estático/DMSA: Todo paciente pediátrico que ha presentado una ITU alta debe realizarse un cintigrama renal a los 6 meses o más desde el episodio para descartar la presencia de cicatrices renales. La presencia de cicatrices obliga a descartar un RVU como factor predisponente a ITU alta.
- Uretrocistografía miccional seriada (UCG): Es el examen de elección para el diagnóstico y la graduación del RVU. Siguiendo al grupo de estudio internacional de reflujo en niños, lo clasifica en 5 tipos (Figura 11). Para optimizar el rendimiento del examen, es importante incluir al menos dos fases miccionales.

- Cintigrafía isotópica directa: En este estudio se instila un radio isótopo intravesical al paciente y posteriormente se obtienen imágenes en la gama cámara durante el llene y la micción. Tiene muy buena sensibilidad y especificidad para la detección de RVU, pero no permite delimitar la anatomía y requiere cateterismo del paciente.
- Cintigrafía Isotópica Indirecta: Examen de elección para el seguimiento de la persistencia del RVU en pacientes con control de esfínter. Tiene la ventaja de utilizar el llene natural de la vejiga por lo que no requiere cateterismo vesical, sin embargo, sólo se puede realizar en pacientes mayores que colaboren con el examen. Tiene una sensibilidad menor que la UCG (75%) y una especificidad mayor al 90%.
Tratamiento
El objetivo del tratamiento del RVU es prevenir las recurrencias de las infecciones urinarias febriles y reducir el riesgo de daño renal. Por ende, el tratamiento del reflujo en si mismo no está indicado, pues sabemos que el paso de orina estéril al sistema colector renal no provoca infecciones ni daño renal.
El concepto más importante es que el tratamiento del RVU debe ser individualizado, considerando: forma de presentación (sintomático o asintomático), edad del paciente, control de esfínter, presencia o no de BBD, presencia/no circuncisión, presencia/no de cicatrices renales, adherencia al tratamiento y preferencia del paciente y/o sus cuidadores. El tratamiento será lo más conservador en la mayor parte de los pacientes. Es importante comprender que mientras no exista colonización vesical por uropatógenos, no existirán problemas asociados al reflujo subyacente. Por otra parte, sabemos que la historia natural del reflujo, en sus distintos grados, es hacia la resolución espontánea. En la Figura 12 vemos la tasa de mejoría del RVU durante la infancia.
Fig. 12. Probabilidad de resolución del RVU (Tullus, 2014)
Tratamiento
Las modalidades de tratamiento incluyen:
- Prevención de la recurrencia de las infecciones bajas: Es el punto de partida del daño renal en pacientes con RVU. Si evitamos la colonización bacteriana del tracto urinario inferior, no habrá problemas con el RVU. Las estrategias de tratamiento incluyen: profilaxis antibiótica, tratamiento de la disfunción vesical e intestinal y/o circuncisión
- Corrección quirúrgica del RVU (modalidades endoscópica, laparoscópica, robótica y abierta): Es motivo de debate el beneficio de la corrección quirúrgica del reflujo y es muy importante señalar que siempre debe ser en conjunto con un manejo agresivo del componente vesical del RVU. Indicaciones relativas de cirugía incluyen: presencia de alteraciones anatómicas como divertículo para ureteral y/o doble sistema renal, deterioro de la función renal, mala adherencia al tratamiento médico y/o infecciones en pacientes en profilaxis antibiótica, reflujo de grado alto en pacientes mayores de 3-4 años.
- El tratamiento endoscópico del RVU resulta ser una alternativa atractiva por tratarse de un procedimiento ambulatorio con alta tasa de éxito (85%). Hay que considerar que se pueden presentar complicaciones entre un 10-15%, la más frecuente es el RVU persistente. Hasta un 5% de los niños puede evolucionar con obstrucción del uréter distal post inyección, por lo que se requiere un seguimiento estricto de los pacientes tratados por esta vía.
- El tratamiento quirúrgico sigue siendo el “gold standard” en el tratamiento del RVU. Las técnicas clásicas de Cohen y Gregoir tienen una alta tasa de éxito (98%) y una baja tasa de complicaciones (1-2%). Estas técnicas pueden y han sido replicadas en cirugía laparoscópica y robótica.
Tumores Genitourinarios Pediátricos
Tumores Testiculares
En Chile, los tumores testiculares pediátricos representan anualmente menos del 10% de los casos nuevos de tumores solidos de la infancia, es decir menos de 15 casos nuevos a nivel nacional13. A nivel internacional se estima una incidencia entre 0.5 a 2 casos por 100.000 niños menores de 15 años14,15. La mayoría de los tumores testiculares de la infancia aparecen en pacientes menores de dos años y la presentación clínica más frecuente es la presencia de un aumento de volumen testicular indoloro16. En ocasiones puede existir el antecedente de trauma, dolor o hidrocele. El examen físico revela una masa testicular dura, indolora con el consecuente aumento de volumen escrotal unilateral. La transiluminación puede ser útil para distinguir componentes sólidos o quísticos. Otra forma de presentación clínica es la pubertad precoz. Los diagnósticos diferenciales incluyen la torsión testicular, hernia inguino escrotal, epididimitis y el hidrocele15.
Los tumores testiculares se clasifican de acuerdo con su histología en dos grupos principales, los tumores de células germinales y tumores estromales. El primer grupo es el más frecuente, representando alrededor del 80% de los tumores testiculares pediátricos e incluye los carcinomas embrionarios infantiles, tumor de saco vitelino, los teratomas y los quistes epidérmicos o tumores dermoides. Los tumores estromales comprenden los tumores de células de Leydig, los tumores de células de Sertoli, los tumores de las células granulosas y los tumores estromales mixtos. El seminoma, un tipo de tumor de células germinales, que representan el 89% de los tumores testiculares en adultos, rara vez se encuentra en pacientes pediátricos16.
| LINEA CELULAR | TIPOS |
| Células germinales |
|
| Estromales |
|
| Otros tumores primarios testiculares |
|
| Tumores Para testiculares |
|
| Secundarios |
|
Tabla 4. Clasificación de los tumores testiculares pre puberales
Teratoma Maduro: Los teratomas maduros son los tumores testiculares más frecuentes y tienen un comportamiento biológico benigno. Histológicamente presentan ADN diploide con presencia de los 3 tipos celulares embrionarios (ectodermo, mesodermo y endodermo). Dada su naturaleza benigna, probablemente son menos reportados que otras formas histológicas.
Teratoma Inmaduro: Son infrecuentes y tienen comportamiento maligno potencial. Tienen bajo riesgo de recaída, respondiendo efectivamente a la quimioterapia. Existen reportes aislados de metástasis secundarias a teratomas testiculares inmaduros15.
Tumor de saco vitelino o del seno endodérmico: Es considerado el tumor maligno más frecuente en el grupo pediátrico. Generalmente se presenta en niños menores de dos años y tiene potencial de metástasis a pulmones y linfonodos retroperitoneales. De acuerdo con el registro de tumores de la Academia Americana de pediatría, cerca del 80% de ellos son diagnosticados en etapa I14. Este tumor presenta elevación de la alfa-feto proteína en el 90% de los casos, que debe ser monitorizada previo a la orquidectomía y posterior a ella, como marcador de presencia de tumor residual. No se debe olvidar la elevación fisiológica de la alfa-feto proteína del recién nacido y durante el primer año de vida de modo de no interpretar en forma incorrecta un resultado elevado en una lesión testicular probablemente benigna16. El pronóstico es excelente, con una sobrevida mayor al 90% incluso en pacientes con tumor residual o metástasis, dada la sensibilidad del tumor a la quimioterapia.
Estudio
El estudio de una masa testicular contempla exámenes de laboratorio e imágenes. Dado el alto porcentaje de lesiones benignas que se presentan como una masa testicular, es importante diferir el estudio de diseminación en aquellos pacientes en que se ha confirmado histológicamente la naturaleza maligna de la lesión o en aquellos con una fuerte sospecha de patología tumoral maligna15.
- Alfa feto proteína: Esta cadena polipeptidica de aminoácidos es producida por el saco embrionario, hígado e intestino fetal. Su vida media es de aproximadamente 5 a 7 días y sus valores deben ser ajustados por la edad del paciente ya que normalmente el valor adulto no se alcanza sino hasta los 8 meses de vida. Característicamente, la alfa-feto proteína está elevada en los tumores del seno endodérmico donde se encuentra sobre su valor normal en cerca del 90% de los casos. Con menor frecuencia, los pacientes con teratomas también pueden presentar una elevación de esta proteína.
- Gonadotrofina coriónica humana-b: Esta glicoproteína tiene una vida media de 24 horas y rara vez esta presente en tumores testiculares en preadolescentes. En el reporte de la Academia Americana de Pediatría del año 2002, ninguno de los pacientes evaluados por tumores testiculares presentó elevación de este marcador3. En ocasiones los pacientes con teratomas y gonadoblastomas pueden presentar elevación de esta glicoproteína, pero no así aquellos que presentan un tumor del seno endodérmico. Su elevación es característica en tumores tipo adulto tales como carcinoma embrionario o coriocarcinoma.
- Testosterona: En un paciente con pubertad precoz y sospecha de un tumor de células de Leydig se debe solicitar el estudio de los niveles de testosterona, los que en ocasiones se encuentran anormalmente altos en tumores hormonalmente activos.
- Ecografía: Frente a la sospecha diagnostica es el examen de elección. Permite distinguir tumores testiculares de lesiones no tumorales y la ubicación intra o extra testicular de ellas. Las imágenes ecográficas también pueden ser sugerentes de un tipo específico de lesión. Especialmente útil es la visualización de lesiones quísticas simples de carácter benigno o la presencia de calcificaciones gruesas más frecuentes en teratomas. Este examen no permite el diagnostico definitivo de una lesión maligna, pero es clave para planificar el tratamiento.
- Estudio de diseminación: La tomografía axial computada de abdomen está indicada sólo frente a la sospecha de tumores malignos tales como lesiones extra testiculares tipo rabdomiosarcoma para testicular, o frente a un paciente mayor de 6 meses con una lesión testicular y elevación significativa de la alfa-feto proteína en la que sospechamos un tumor del seno endodérmico. En ambos casos es perentorio el estudio de diseminación para planificar una terapia adyuvante acorde a los hallazgos de las imágenes. Asimismo, frente a cualquiera de estos dos escenarios clínicos, se debe tomar una radiografía de tórax y/o una tomografía de tórax en la búsqueda de metástasis pulmonares17.
Tratamiento
El tratamiento de los tumores testiculares es quirúrgico. Tanto en sospecha de lesiones benignas como malignas el abordaje es por vía inguinal, asegurando una incisión amplia que permita la exteriorización del testículo sin manipulación excesiva. Frente al diagnostico de masa testicular, con exámenes de laboratorio normales, es muy probable que la lesión tenga un carácter benigno. Frente a este escenario, se recomienda la extirpación de la masa tumoral preservando el tejido testicular normal. La técnica incluye el control vascular por vía inguinal con una cintilla vascular lo más cercano al orificio inguinal profundo. Posteriormente se realiza una biopsia excisional rápida y una vez confirmado el diagnostico de un teratoma maduro, de un quiste epidérmico o de un tumor de células de Leydig se procede a la preservación del tejido testicular normal, removiendo sólo el tumor. Es fundamental confirmar la presencia de tejido testicular normal alrededor de la muestra, especialmente en niños que se acercan a la pubertad en cuyos casos existe mayor riesgo de malignización de teratomas17.
En el caso de elevación de alfa feto proteína, es altamente probable que sea un tumor tipo del seno endodérmico y en estos casos está indicada una orquidectomia radical por vía inguinal. Mas del 90% de estos tumores se encuentran confinados al testículo y este procedimiento será curativo sin requerir otras intervenciones. Los tumores del seno endodérmicos son muy sensibles a la quimioterapia basada en Cisplatino por lo que rara vez se indicará una linfadenectomía radical retroperitoneal en caso de enfermedad residual. La figura 13 muestra el aspecto al examen físico, ecográfico e intraoperatorio de un tumor testicular maligno (seno endodérmico)

Pronóstico
El pronóstico en general de las masas tumorales testiculares en la edad pediátrica es excelente, incluso en lesiones malignas o en presencia de metástasis. La estadística nacional publicada en 2004 por el programa nacional de drogas antineoplasticas infantiles (PINDA), que incluyó 29 pacientes tratados en el período comprendido entre 1993 y 1999 la sobrevida total alcanzo un 96% con una mediana de seguimiento de 40 meses. Del total de pacientes tratados, hubo 8 de ellos que presentaron recaída local o a linfonodos retroperitoneales, ningún paciente recayó con metástasis a distancia17.
Tumor de Wilms
Los tumores renales representan cerca del 6% de los tumores malignos de la infancia y de estos, sobre el 90%, son nefroblastomas o tumor de Wilms18-19. La edad de presentación más frecuente es entre los 2 y 3 años, con una incidencia uniforme en distintas partes del mundo3. La forma de presentación clínica habitual es la presencia de una masa abdominal indolora que puede ser detectada por familiares o en un examen de rutina de salud. Hasta un 25% de los pacientes pueden manifestar hematuria, dolor abdominal y/o baja de peso. Frente una masa abdominal en un niño, los diagnósticos diferenciales incluyen patologías benignas tales como: patología benigna renal (hidronefrosis, displasia renal multiquística), quistes de origen digestivo (duplicación intestinal, quiste de colédoco, quistes mesentéricos) y/o ginecológicos; así como también tumores malignos, como el neuroblastoma, hepatoblastoma, linfoma y tumores de células germinales.
El examen físico de un paciente portador de un tumor de Wilms revelará la presencia de una masa abdominal unilateral, indolora. Los sitios de metástasis más comunes del tumor de Wilms son los linfonodos abdominales, seguido por nódulos pulmonares y en menor frecuencia, hepáticos. Todos ellos se presentan asintomáticamente, por lo que incluso en etapas avanzadas de la enfermedad los pacientes presentan pocos síntomas. Sólo un 5% de los pacientes tendrán una presentación bilateral.
Aproximadamente un 12% de los pacientes portadores de un tumor de Wilms pueden presentar una forma sindromática, muchos de ellos debido a mutaciones del gen WT1 que está asociado a la embriogénesis del sistema urinario. Entre estos destacan el síndrome de WAGR (Tumor de Wilms, Aniridia, anomalías Genitourinarias, Retraso mental) y el síndrome de Denys-Drash (asociado a insuficiencia renal precoz, tumor de Wilms y disgenesia gonadal). También existe asociación con el síndrome de Beckwith-Wiedemann (hemi hipertrofia) el que está presente en el 3% de los pacientes portadores de un tumor de Wilms20. Frente a la presencia de hemi hipertrofia, aniridia o la confirmación de la mutación del gen WT1 se recomienda realizar tamizaje periódico para descartar la presencia de un tumor de Wilms.
Estudio
Frente a la sospecha de una masa abdominal en pediatría, es de elección la ecografía de abdomen. Esta mostrará una masa renal solida uni o bilateral. Posteriormente, se complementará el estudio con una tomografía axial de abdomen, pelvis y tórax, para evaluar la extensión tumoral, la apariencia de los linfonodos y la presencia de metástasis hepáticas y/o pulmonares. Frente a sospecha de compromiso tumoral vascular (vena cava inferior) o lesiones tumorales atípicas se puede solicitar una resonancia nuclear magnética como complemento al estudio.
La figura 14 muestra una resonancia y el aspecto intraoperatorio de un paciente portador de un tumor de Wilms derecho

Tratamiento y etapificación:
El tratamiento del tumor de Wilms tiene tres pilares: Quimioterapia, Cirugía y Radioterapia. Desde el año 2019, nuestro país sigue el protocolo de tratamiento UMBRELLA, que surge con el objeto de mejorar la sobrevida en aquellos subgrupos de peor pronóstico o con recaídas frecuentes y es una fusión del protocolo Norte Americano (RTSG) y Europeo (SIOP)18. De esta forma, la mayor parte de los pacientes con sospecha de tumor de Wilms por clínica y estudio imagenológico compatible, irán a quimioterapia preoperatoria de reducción (entre 4 a 6 semanas) y posteriormente a cirugía.
La cirugía estándar en pacientes portadores de un tumor de Wilms unilateral es la nefrectomía radical abierta, junto con la resección de linfonodos retroperitoneales. Existe experiencia limitada en tumores de volumen reducido con resección por laparoscopía, así como la técnica de Neprhon-sapring o nefrectomías parciales, principalmente indicada en tumores bilaterales al diagnóstico. La etapificación y el tratamiento complementario se realizará posterior a la cirugía basado en los hallazgos quirúrgicos, estado de los linfonodos y tipo histológico. La tabla 1 resume las etapas de tumor de Wilms18
| ETAPA | DESCRIPCIÓN |
| 1 | Tumor limitado al riñón o grasa peri renal pero confinado a una pseudo cápsula, sin infiltración de ella ni de las paredes de la pelvis renal ni uréter |
| 2 | Tumor renal con infiltración local de grasa, uréter, vasos sanguíneos y/o linfáticos o tejido adyacente (exceptuando glándula supra renal y linfonodos), resecado completamente (márgenes libres) |
| 3 | Tumor con infiltración local sin márgenes libres, linfonodos positivos, compromiso adrenal, ruptura tumoral, implantes tumorales (superficie peritoneal o a distancia) |
| 4 | Metástasis a distancia hematógenas (hígado, pulmón, cerebro) o linfonodos positivos extra abdominales |
| 5 | Tumor bilateral al diagnóstico |
Tabla 5 (Adaptado de Vujanic GM., Gessler M., Ooms AHA et al, 2017)
Adicionalmente se estudiarán algunos biomarcadores asociados a peor pronóstico, siendo la ganancia del cromosoma 1q la más frecuente. La radioterapia está indicada en pacientes con criterios de etapa III y aquellos con histología desfavorable (anaplasia), desde la etapa II21.
Pronóstico
La sobrevida de los pacientes portadores de un tumor de Wilms es excelente, para los pacientes en etapa I y II, con histología favorable, se estima sobre el 85-90%. En pacientes sometidos a radioterapia en Chile, la sobrevida a 4 años fue de un 78%20. Los pacientes sometidos a tratamiento por un tumor de Wilms, deben continuar en seguimiento para la detección precoz de recaídas, presencia de tumor contralateral metacrónico y la aparición de insuficiencia renal.
Rabdomiosarcoma genitourinario
En la población pediátrica, el rabdomiosarcoma (RMS) es el sarcoma de tejidos blandos más frecuente y aproximadamente un 20% de ellos tienen su origen en el sistema genitourinario22. En Chile, el RMS representa el cuarto tumor sólido más frecuente en la infancia23. Los dos tipos histológicos, el rabdomiosacroma embrionario (RMSE) y el alveolar (RMSA) tienen un comportamiento biológico distinto, en términos de edad de presentación, sitio primario, propensión a metástasis y pronóstico24. Las ubicaciones genitourinarias que pueden verse afectadas por un RMS incluyen: tracto genital femenino (vagina, útero y cérvix), vejiga y próstata. El sitio más común es el vésico/protático y representa sólo el 5% de todos los RMS22.
La edad media de presentación es alrededor de los 5 años y frecuentemente se manifiesta como hematuria, disuria y/o retención urinaria. Frente a la presencia de un cuadro de retención urinaria aguda en un niño previamente asintomático, es mandatorio descartar esta patología.
Los diagnósticos diferenciales incluyen patología benigna tales como infección del tracto urinario, litiasis y uretritis inespecífica. Al examen físico puede ser evidente la presencia una masa abdominal y en niñas se puede observar la protrusión tumoral en el introito vaginal. Como se ve en la figura 15, es característica la imagen de protrusión tumoral en racimo de uva del RMSE variedad botroide.
Estudio y etapificación
Frente a la sospecha clínica el estudio se iniciará con una ecotomografía de abdomen y pelvis. La confirmación de una masa tumoral de origen genitourinario llevará a complementar con una tomografía axial (TAC) de abdomen y pelvis y/o una resonancia nuclear magnética. El estudio de diseminación contempla la TAC de tórax y el cintigrama óseo. Recientemente, se rescata la utilidad del PET scan como parte del estudio inicial, que permitiría confirmar el compromiso de médula ósea, huesos, linfonodos y tejidos blandos22. Los RMS se clasifican en tres grupos de riesgos: bajo, intermedio y alto. Esta división permite racionalizar el tratamiento, pero incluyen múltiples elementos tales como localización, tamaño tumoral, estado ganglionar, histología (embrionario o alveolar), presencia de metástasis y resección quirúrgica (completa/incompleta). La tabla 5 resume la etapificación y la tabla 6 como clasificar los RMS genitourinarios.
| ETAPA | DESCRIPCIÓN |
| I | Localización en genitales femeninos o para testicular (Cualquier tamaño y/o nodos) |
| II | Localizado en Vejiga/Próstata, menor a 5 cm, nodos negativos o no evaluados |
| III | Localizado en Vejiga/Próstata, menor a 5 cm, nodos positivos
Localizado en Vejiga/Próstata, mayor a 5 cm, nodos negativos/no evaluados |
| IV | Cualquier localización, tamaño y estado nodal, con metástasis a distancia |
Tabla 5 (Adaptado de Saltzman y Cost, 2018)
| RIESGO | ETAPA/RESECCIÓN/HISTOLOGÍA |
| BAJO |
|
| INTERMEDIO |
|
| ALTO |
|
Tabla 6. Estratificación de riesgo según etapa, resección e histología
Tratamiento
El tratamiento del RMS se apoya en la cirugía, radioterapia y quimioterapia. Dado el comportamiento biológico agresivo y la baja sobrevida de los pacientes, la resección tumoral radical era lo históricamente recomendado. Sin embargo, en la actualidad, el tratamiento a virado a la preservación de órganos y la radioterapia ha tomado un rol protagónico, estando indicada en cerca del 90% de los pacientes23.
La quimioterapia preoperatoria está indicada sólo en casos seleccionados y siempre que sea posible la resección tumoral completa con morbilidad aceptable, esta debe ser ofrecida. Frente a tumores de mayor tamaño en la que la resección completa implica una morbilidad importante, como por ejemplo una cistectomía, es preferible la resección tumoral parcial, seguida por radioterapia. Con la finalidad de reducir los efectos adversos asociados a la radioterapia abdominopélvica, existe experiencia tanto nacional como internacional en el uso de braquiterapia como terapia complementaria en resecciones incompletas con el fin de preservar la función vesical 22,23.
Otras terapias en desarrollo incluyen la terapia de protones, una modalidad de radioterapia alineada en el mismo objetivo que la braquiterapia, la de reducir la toxicidad y minimizar la radiación en órganos circundantes al tejido tumoral 22. Estas terapias han demostrado, en estudios iniciales, ser equivalentes en sobrevida y eventos libres de enfermedad a la radioterapia convencional, pero con menor morbilidad.
Pronóstico
La estadística PINDA del año 2000 en Chile, estimó la sobrevida global a 5 años en un 55%23. Según grupo de riesgo y utilizando los nuevos protocolos de etapificación, la literatura internacional cifra en aproximadamente un 80% la sobrevida en el grupo de bajo riesgo, entre un 50-70% para riesgo intermedio y en menos de un 30% para pacientes de alto riesgo22.
Bibliografía
- Thomas, D. F. M.; Duffy, Patrick G.; Rickwood, A. M. K. (2008): Essentials of paediatric urology. 2nd ed. /. London, Boca Raton, FL: Informa Healthcare; Distributed in North America by Taylor & Francis.
- Ashcraft, Keith W.; Holcomb, George W.; Murphy, J. Patrick; Ostlie, Daniel J. (2010): Ashcraft’s pediatric surgery. 5th ed. Philadelphia: Saunders/Elsevier.
- I.A. Hughes et al. Consensus statement on management of intersex disorders. J Ped Urology (2006) 2, 148-162.
- Cools M. et al. Consensus Statement: Caring for individuals with a difference of sex development (DSD). Nat Rev Endocrinol (2018) 14, 415 – 429.
- Lopez P.J. et al. Manual de Cirugía Pediátrica de la A a la Z. Capitulo Trastorno del Desarrollo Sexual (2019), 369 – 372.
- Campbell, Meredith F.; Walsh, Patrick C.; Retik, Alan B.; Vaughan, E. Darracott; Wein, Alan J. (2002): Campbell’s urology. 8th ed. Philadelphia, PA: Saunders.
- Gearhart, John P.; Rink, Richard C.; Mouriquand, Pierre D. E. (2010): Pediatric urology. 2nd. Philadelphia: Saunders/Elsevier.
- Tekgul S., Nijman RJM., Hoebeke P., Canning D., Bower W., Von Gontard A. Diagnosis and Management of Urinary Incontinence in Chilhood. International Continence Society. Retrived from https://www.ics.org/Publications/ICI_4/files-book/Comite-9.pdf
- Austin PF., Bauer SB., Bower W. et al The standardization of terminology of lower urinary tract function in children and adolescents: update report from the Standardiza tion Committee of the International Children’s Continence Society. J Urol 2014 Jun;191(6):1863-1865
- Tullus K., Vesicourteric Reflux in Children. Lancet. 2015 Jan 24;385(9965):371-9
- Nelson C. et al. Incidence of urinary tract infection among siblings of children with vesicoureteral reflux. Acad Pediatr. 2016 July ; 16(5): 489–495
- Sepulveda JA. Reflujo vesicoureteral. Manual de Cirugía Pediátrica, Sociedad Chilena de cirugía Pediátrica, 2019. https://www.schcp.cl/wp-content/uploads/2019/10/Manual-de-Cirug%C3%ADa-Pedi%C3%A1trica-de-la-A-a-la-Z-SChCP-2019.pdf
- Vargas L. Cáncer en pediatría. Aspectos generales. 2000;7(4):283-295
- Ross J., Kay R. Prepubertal Testis Tumors. Rev Urol. 2004;6(1):11-18
- Chung J., Lee S. Overview of Pediatric Testicular Tumors in Korea. Korean J Urol. 2014;55(12):789-796
- Zamilpa I., Koyle M. Pediatric Testicular Tumors. Fundamentals of Pediatric Surgery. Springer, USA. 2010. Chapter 97. pp 749-754
- García H., Sepúlveda L., Tordecilla J. Resultados Protocolo 1993-1999 para tumores testiculares germinales no seminoma (PINDA). Rev Chil Pediatr. 2004;75(2):129-138
- Vujanic GM., Gessler M., Ooms AHA et al. The UMBRELLA SIOP_RTSG 2016 Wilms tumour pathology and molecular biology protocol. Nat Rev Urol, Vol 15 ; 743-752, 2017
- González g., Tumores sólidos en niños: diagnóstico y terapéutica quirúrgica. Rev Med Clin Condes 21(1) 120-129, 2010
- Friedman A. Wilms Tumor. Pediat in Rev, 34; 328-330, 2013
- Isa N., Reyes M., Russo M. Resultado del tratamiento del tumor de Wilms en población pediátrica. Rev Chil Pediatr 84(6): 628-633, 2013
- Saltzman A., Cost NG., Current treatment of Pediatric Bladder and Prostate Rhabdomyosarcoma. Curr Urol Rep. (22);19(1):11, 2018
- Malempati S., Hawkins DS., Rhabdomyosarcoma: review of the Children’s Oncology Group (COG) SoftTissue Sarcoma Committee experience and rationale for current COG studies. Pediatr Bood Cancer 15;59(1):5-10, 2012
- Goset K., Córdova A., Varas M., Badínez L., Tratamiento actual del Rabdomiosarcoma pediátrico en Chile. Presentación de 2 casos clínicos. Rev Chil Pediatr, 73(4);375-379, 2002.
